
Abstract
The barocaloric effect (BCE) has emerged as an intense research topic in regard to efficient and clean solid-state refrigeration. Materials with solid-liquid phase transitions (SL-PTs) usually show huge melting entropies but cannot work in full solid-state refrigeration. Here, we report a colossal barocaloric effect realized by exploiting high entropy inherited from huge disorder of liquid phase in amorphous polyethylene glycol (PEG), which is solidified by introducing 5 wt.% polyethylene terephthalate (PET). Transmission electron microscopy (TEM) combined with X-ray diffraction (XRD) demonstrates the amorphous nature of the high-temperature phase after fixation by PET. Although PEG loses its –OH end mobility in amorphous solid, high entropy still retains owing to the retained high degrees of freedom of its molecular chains. The remaining entropy of amorphous PEG is up to 83% of that of liquid PEG in PEG10000/PET15000, and the barocaloric entropy change reaches ΔSp ∼ 416 J·kg−1·K−1 under a low pressure of 0.1 GPa, which exceeds the performance of most other BCE materials. Infrared spectra combined with density function theory (DFT) calculations disclose conformational change from the liquid to amorphous state, which explains the origin of the large entropy retained and hence the colossal BCE of the solidified PEG. This research opens a new avenue for exploring full solid-state barocaloric materials by utilizing genetic high entropy from huge disordering of liquid phases in various materials with SL-PTs.
Similar content being viewed by others


Introduction
Refrigeration technology based on hydrofluorocarbons has produced severe ozonosphere hole and greenhouse effects, which have stimulated the exploration of highly efficient and environmentally friendly alternatives1,2,3. Materials with mechanical responses, which exhibit field-driven caloric effects in the vicinity of phase transitions (elastocaloric effects under uniaxial stress and barocaloric effects (BCE) under hydrostatic pressure), are expected to replace the existing refrigerants1,4,5. BCEs have been achieved for a variety of phase transition materials, including inorganic/organic ferroic materials6,7,8,9,10 and materials undergoing spin crossover11,12. What is remarkable is the discovery of colossal BCEs for plastic crystals13,14, where barocaloric entropy change (ΔSp) reaches 380–690 J·kg−1·K−1, which is regarded as a milestone in the field of BCEs. The origin of this effect is inextricably bound to the change of intermolecular hydrogen bonds and orientational disorder during solid-solid phase transitions (SS-PTs)15,16.
On the other hand, materials undergoing solid crystalline to liquid phase transitions (SL-PTs) usually contain huge latent heats (150–300 J/g), even comparable to those of conventional gas-liquid cooling refrigerants (e.g., Freon-based materials with latent heats of 130–280 J/g), originating from huge disorder of molecular arrangement and fracture of intermolecular hydrogen bonds in the liquid phase, such as paraffins and fatty acids17,18. Typically, Lin et al. reported a colossal and reversible barocaloric effect in n-alkanes with SL-PTs, and the entropy change reached up to ~700 J·kg−1·K−1 at a low pressure of ~50 MPa due to the twisting and random thermal motions of the molecular chains18. However, SL-PT materials are prone to problems, such as liquid leakage from the containers, phase separation caused by the overall convective flow during the phase transition, and supercooling during solidification, which inevitably cause the corrosion damage of refrigeration devices and the irreversible performance of refrigerants19,20. Therefore, it is of great significance to explore the curing of the liquid phase while maintaining huge disorder and high entropy.
Polyethylene glycol (PEG, HO(CH2CH2O)nH)21,22 is a line-type molecule composed of -OH end groups and flexible methylene (CH2) repeating units with a high rotational capacity (Fig. 1a). With increasing temperature, PEG transforms from a crystalline solid into a liquid, accompanied by huge latent heat. The phase transition enthalpy and entropy changes are ~150–190 J/g and 400–600 J·kg−1·K−1, respectively. Considering the -OH terminal groups of PEG, rationally engineering hydrogen bonds may solidify the flow of PEG segments. Polyethylene terephthalate (PET, C10H8O4)m)23,24 is a degradable crystalline saturated polyester composed of rigid aromatic rings, CH2 and polar ester groups (C=O) with exposed oxygen acceptors (Fig. 1b). By introducing PET for copolymerization (Fig. 1c), the terminal –OH (black ellipse) groups at both ends of each PEG chain are prone to connect with the PET chains by forming hydrogen bonds (O–H···O, red ellipse) due to the lone pairs of electrons on the exposed oxygen atoms of PET, which solidifies the flow of PEG segments. Then, the liquid PEG converts into an amorphous solid of PEG/PET. However, although the PEG molecules are bound at both ends, the molecular chains, except the terminal –OH groups, still retain a high rotational/vibrational degree of freedom due to the presence of a large amount of C–C σ-bonds (CH2–CH2), which can make the huge transition entropy change still kept.

a Single PEG molecule, where the black and blue dashed boxes denote the terminal hydroxyl groups (–OH) and the repeating units –CH2–CH2–O–, respectively. b Single PET molecule, where the blue dashed box denotes the repeating unit –C10H8O4–. c PEG/PET, where the molecular chain containing the aromatic ring in the middle is the skeleton chain PET, while the molecular chains around the skeleton are PEG. The black, pink, blue, red dashed ellipses denote the –OH, CH2 groups, C–O, O–H···O bonds, respectively. The purple dots represent the possible hydrogen bonds formed between the oxygen atoms and the terminal –OH groups of PEG. The configuration of line-type C–C σ-bonds in the PEG molecule causes the whole molecule to be highly flexible and has good conformational compliance with the rigid PET molecule so that the shape of the PEG chains can change with the shape of the rigid PET chains. The red, gray, and white balls represent the oxygen, carbon, and hydrogen atoms, respectively, in the PEG and PET molecules.
We successfully synthetized PEG/PET by introducing 5 wt.% PET for copolymerization. Transmission electron microscopy (TEM) combined with X-ray diffraction (XRD) measurements demonstrate the amorphous nature of the high-temperature phase after fixation by PET. The remaining entropy in amorphous PEG is up to 83% that of liquid PEG in PEG10000/PET15000, and the measured barocaloric entropy change exceeds the performance of most other BCE materials. Infrared (IR) spectra combined with density functional theory (DFT) calculations disclose the specific conformational change from the liquid to the amorphous state and explain the origin of the large entropy remaining in amorphous PEG and hence the colossal BCE in the solidified PEG.
Materials and methods
Preparation of PEG10000/PET15000
First, the raw materials PEG with Mr = 10,000 and PET with Mr = 15,000 were dried, whereby PEG was dried with a silica gel desiccant at room temperature, and PET was placed in a drying oven at 110 °C for more than 4 hours until constant weight. Then, PEG and PET were individually dissolved in DMSO (dimethyl sulfoxide) at mass ratios of 1:3 and 1:20 at 40 °C and 180 °C, respectively. Subsequently, the resulting solutions of PEG and PET were mixed at 180–200 °C and formed a thick paste quickly, which was then washed and filtered after cooling to room temperature. After filtering, the samples were placed in a vacuum drying oven at ~100 °C for 24 hours, and then PEG10000/PET15000 was obtained.
Characterizations
XRD measurements were carried out on a powder sample using a Rigaku-SmartLab diffractometer with Cu-Kα = 1.5406 Å radiation over 10°–60° with a constant step of 0.02°, which provided variable temperature from 4 K to 1000 K. TEM and selected area electron diffraction (SAED) measurements were carried out by a JEOL JEM-2100F TEM with an accelerating voltage of 200 kV for specimens placed on copper grids, which provided temperature environments from 100 K to 300 K. The low temperature was realized by liquid nitrogen, and the testing process was carried out in heating mode.
Variable temperature IR spectra were collected in the region from 1000 cm−1 to 3800 cm−1 by a BRUKER TENSOR FTIR (Fourier Transform Infrared) spectrometer, which provided temperature environments from 295 K to 400 K. To avoid signal saturation and obtain a high signal-to-noise ratio for the IR spectra, both single PEG10000 and PEG10000/PET15000 were diluted by KBr powders before measurements. For the convenience of comparison, the same initial temperature of 295 K was selected despite the different transition temperatures (Tt) for PEG10000 (339 K) and PEG10000/PET15000 (333 K), and the temperature increments around Tt were both 2 K.
Heat flow measurements were performed using a commercial high-pressure differential scanning calorimeter (μDSC7 evo microcalorimeter from SETARAM). A temperature ramping rate of 1 K/min was adopted for both the heating and cooling processes. Hydrostatic pressure ranging from atmospheric pressure to 0.1 GPa was applied by compressed N2 gas with high purity (99.999%).
Density functional theory (DFT) calculations
Based on the Gaussian09 software package, quantum chemical calculations of the IR spectra of PEG molecules with free (denoted as free-PEG) and fixed –OH groups at both ends (denoted as fixed-PEG) were performed using density functional theory (DFT). For simplicity, seven repeating units (–CH2CH2O–) of a PEG molecule were selected, and the B3LYP function of the 6–311G* basis set was used to optimize the molecular structure and calculate the IR vibrational frequencies. Polarization functions were used to well optimize the structure and frequency accuracy, and the corresponding frequency correction factor was 0.97305 during calculation, while the convergence criterion was set at 10−6 eV.
For the crystalline state, the atomic positions of the PEG crystalline state used for IR spectral calculations were derived from the optimal molecular model by minimizing the energy of the TTT and TGT conformations, where the molecular chain changed slightly under the energy convergence criterion. Then, the IR spectra of the low-temperature crystalline state for fixed-PEG were calculated by constraining the coordinates of –OH groups at both ends, and those of the free-PEG without limitation were calculated as well.
The possible conformations of the liquid/amorphous state were further calculated for free-PEG and fixed-PEG. Six possible conformations of TGT, TGG, GGG, TTT, GTG, TTG were considered for both free- and fixed-PEG during the calculations, and the atomic positions used for the IR spectra calculations were derived from the optimal molecular model by minimizing the energy of the 6 conformations.
Methods for obtaining the barocaloric entropy change (ΔSp) and adiabatic temperature change (ΔTp)
A quasi-direct method was used to measure barocaloric performance1,9. DSC equipped with a μDSC7 evo microcalorimeter from SETARAM was employed to measure the isobaric heat flow curves at different pressures in the range 0–0.1 GPa. Moreover, the specific heat capacity Cp at ambient pressure was also measured using the same calorimeter. Entropy as a function of temperature (S-T curve) at different pressures is given by the following formula1,18,
where \(\frac{{dQ\left( {T,P} \right)}}{{dt}}\) is the heat flow and \(\frac{{dT}}{{dt}}\) is the temperature sweep rate. The measured heat flow after subtracting the baseline was used when calculating entropy, and Cp(T, P) was approximated as Cp(T) under ambient pressure (see details given in supporting information, SI-4). Then, the barocaloric entropy change (ΔSp) and adiabatic temperature change (ΔTp) were obtained by using the equations ΔSp (T, P1 → P2) = S (T, P2) − S (T, P1) and ΔTp (S, P1 → P2) = T (S, P2) − T (S, P1), where the atmospheric pressure was taken as the initial and end pressures for the compression and decompression processes, respectively. Moreover, the reversible entropy change (ΔSrev) was reckoned as the overlapping of ΔSp during compression and decompression, while similarly, the reversible adiabatic temperature change (ΔTrev) was obtained, excluding the influence of thermal hysteresis14.
Results and discussion
PEG10000/PET15000 was synthesized by reacting the raw materials PEG (HO(CH2CH2O)nH, n ≈ 227) with a relative molecular mass of Mr = 10,000 and PET ((C10H8O4)m, m ≈ 78, Mr = 15,000) in dimethyl sulfoxide (DMSO). The mass ratio was PEG:PET = 95:5, and the corresponding molar ratio was 28.5:1. To observe the morphological changes with temperature, single PEG10000, single PET15000 and PEG10000/PET15000 were heated simultaneously. As shown in Fig. S1, PEG10000/PET15000 remained in a shape-invariant solid-state throughout the entire heating process, while PEG transformed from solid to liquid around its transition temperature (Tt). That is, the liquid PEG successfully converted into a solid in PEG/PET (SI-1). Tt was identified at 340.8 K and 334.6 K upon heating for single PET15000 and PEG10000/PET15000, respectively, from differential scanning calorimetry (DSC) measurements (see details thereafter).
We performed XRD measurements below and above Tt for PEG10000/PET15000, where single PEG10000 and PET15000 collected at room temperature were also measured for comparison (Fig. 2a). For the crystalline state at 300 K, the feature peaks of single PEG10000 were observed for PEG10000/PET15000 with almost the same peak positions and shapes, suggesting an unchanged crystal structure of PEG10000 in PEG10000/PET15000 even 5 wt.% PET was incorporated. At 353 K above Tt, the XRD pattern of PEG10000/PET15000 became a broadened reflection, on which the very small peaks were consistent with the PET reflections. This result implies that the PEG10000 in PEG10000/PET15000 undergoes a transition from crystalline to amorphous solid.

a XRD patterns of PEG10000/PET15000 collected at 300 K and 353 K, and single PEG10000 and PET15000 at 300 K were also measured for comparison. For PEG10000/PET15000, TEM morphologies at b, c 110 K and e, f 300 K environmental temperatures. Panels d and g show the SAED images of the red circled areas in c and f, respectively. h–k SAEDs of different regions in e and f. The dashed blue line marking the same areas in c and f acts as a visual guide.
To further understand this phase transition, TEM studies were carried out on PEG10000/PET15000 at low temperature (110 K) and high temperature (300 K), where an accelerating voltage of 200 kV was used during observations. Figure 2b–g show the TEM morphology and selected area electron diffraction (SAED) at low and high temperatures, respectively. First, it can be seen that the thinnest region at the edge shrank and curled upon heating (red circled region, Fig. 2c, f), suggesting the softening of mechanical properties. Moreover, the corresponding SAED of the edge area at low temperature features sharp and clear diffraction spots (Fig. 2d), indicating an ordered arrangement of crystallinity. However, the SAED of the same area becomes a broadening diffraction halo upon heating (Fig. 2g), a typical diffraction pattern of an amorphous phase. This confirmed the ordered-crystalline to disordered-amorphous transition upon heating under electron beam illumination. Although the environmental temperature (300 K) of TEM observation was lower than the transition temperature (Tt ∼ 334.6 K) of PEG10000/PET15000, thermal fluctuation caused by prolonged electron beam irradiation25,26,27 on the thinnest edge region of organics can drive the transition from crystalline to amorphous solid. Figure 2h–k shows the SAED images of different regions of PEG10000/PET15000 at an environment temperature of 300 K. Due to the nonuniform thickness of the specimen, the same irradiation of the electron beam was unable to convert the entire illuminated area into an amorphous structure; instead, long-range ordered (Fig. 2h, i), short-range ordered (Fig. 2j), and disordered (Fig. 2k) information appeared in different thickness regions.
DSC was used to measure the heat flow for single PEG, single PET, and PEG/PET at atmospheric pressure (the inset of Fig. 3a, b). No phase transition occurred for single PET (Fig. 3b), while both single PEG and PEG/PET showed notable endothermic and exothermic peaks, indicating the occurrence of a first-order phase transition. The Tt of single PEG10000 upon heating/cooling was identified at 340.8 K/324.0 K, while the entropy change (ΔStr) during the SL-PT was calculated to be 543 J·kg−1·K−1. When PEG was fixed on PET, Tt slightly shifted to 334.6 K/313.0 K, while the entropy of the amorphous solid still maintained a great amplitude despite a partial loss. The ΔStr during the SS-PT for PEG10000/PET15000 was 426 J·kg−1·K−1 upon heating. After deducting the influence of 5 wt.% PET that did not contribute to ΔStr, the retained entropy of amorphous PEG was ~83% that of its liquid state (Fig. 3a). The entropy loss can be attributed to the conformational change from a liquid to an amorphous solid for PEG.

a Comparison of the entropy change across Tt for single PEG10000 and PEG10000/PET15000, where the insets show the corresponding heat flow curves after deducting baselines. b Heat flow vs. temperature of single PET. The DSC measurements were performed at a temperature ramping rate of 1 K/min.
To reveal the origin of the high entropy remaining in the amorphous solid, the evolution of the O–H, CH2, C–O, and C–C functional groups/bonds in single PEG10000 and PEG10000/PET15000, as well as the hydrogen bond (O–H···O) connecting PEG10000 and PET15000 (Fig. 1c), were comparatively studied by variable temperature IR spectroscopy (Fig. 4a, b). For the low-temperature crystalline state, the vibration bands reflecting the information of each functional group/bond of the single PEG10000 all appeared for PEG10000/PET15000 (Fig. S2) with almost unchanged vibration frequencies (295 K, Fig. 4a, b), implying that the conformations of the PEG molecular chain in PEG10000/PET15000 remained nearly the same as those of single PEG10000 in the crystalline state. The complete IR spectra are shown in Figs. S3, S4, and the assignments of bands are listed in Table S1.

IR of a single PEG10000 and b PEG10000/PET15000 covering transition temperature Tt. For the crystalline state at 295 K, the vibration bands of each functional group/bond of single PEG10000 all appeared in PEG10000/PET15000 with almost unchanged vibration frequencies, implying that the conformations of the PEG chain in PEG10000/PET15000 remained nearly the same as those of single PEG in the crystalline state. The main difference appears between the liquid and amorphous states above Tt. The evolution of specific bands of primary CH2 groups over a temperature range including Tt for c single PEG10000 and d PEG10000/PET15000. The band at 1022 cm−1 (b) is attributed to the aromatic C–H deformation vibration of PET, which remains unchanged across the phase transition of PEG.
The vibration bands of 3700–3150 cm−1 (Fig. 4a, b, purple zone) were mainly attributed to the end –OH groups of the PEG chain and hydrogen bonds (O–H···O). Due to the long molecular chain (n ∼ 227) of PEG10000 (HO(CH2CH2O)nH), the actual fraction of –OH groups at ends is very low, resulting in the relatively weak vibration information of terminated O–H for single PEG, which remains weak even at the crystalline state (295 K, Fig. 4a). In contrast, PEG10000/PET15000 at the same temperature as the crystalline state (295 K, Fig. 4b) shows a broadened band with a significantly increased intensity, suggesting that the strong hydrogen bond interactions (O–H···O, red ellipse in Fig. 1c) prevailed between the terminal –OH of PEG and the acceptors of PET28,29. (see details in SI-2.1)
For the primary CH2 groups of the PEG chains, the bands at 2888 and 2865 cm−1 are attributed to their symmetric stretching vibrations (denoted as \({\upnu}_{{\mathrm{sCH}}_{2}}\))30. A small but similar blueshift from 2865 to 2869 cm−1 occurred during the SL-PT and SS-PT for both single PEG10000 and PEG10000/PET15000 (Fig. 4a, b, black track line). The band at 2888 cm−1 underwent a large blueshift to 2904 cm−1, and simultaneously, the surrounding area was significantly widened (SI-2.2). Such evolution from the split vibration modes of CH2 into multimode vibrations with continuous frequencies is indicative of a highly disordered molecular arrangement caused by molecular rotation, twisting, or folding in the liquid/amorphous state31. This is also indicative of high entropy in either the liquid state of single PEG or the amorphous state of PEG/PET (see Fig. 3a).
Analogously, the CH2 scissoring vibrations (denoted as \({\updelta}_{{\mathrm{CH}}_{2}}\)) at 1469 and 1455 cm−1 from the internal rotation gauche (G) conformation similarly evolve into a wide band with the occurrence of the SL-PT and SS-PT (Fig. 4a, b, gray zone) for single PEG and PEG/PET29,30,32. The only difference is that the new trans conformation (T, yellow dots in gray area, Fig. 4a) formed at 1485 cm−1 in PEG liquid became blurred and unrecognizable for the amorphous state of PEG/PET (Fig. 4b), implying that the rotation of the molecular chain in amorphous PEG became more difficult than that in liquid PEG, and the new trans conformation did not easily form, contributing to the entropy loss of the amorphous solid (Fig. 3a). In addition, two bands at 1361 and 1344 cm−1 were identified as the wagging vibrations of CH2 (denoted as \({\upomega}_{{\mathrm{CH}}_{2}}\)) relative to the gauche conformation below Tt. Similar evolutions appeared for both single PEG and PEG/PET (Fig. 4c, d). That is, the two merged into one single band (Fig. 4a, b, yellow and purple track lines), which implies that PEG-PEG interchain interactions weakened or even disappeared in the liquid or amorphous state33. Moreover, a new \({\upomega}_{{\mathrm{CH}}_{2}}\) from the CH2–CH2 trans conformation with a feature band at 1324 cm−1 appeared for liquid PEG (Fig. 4a, green zone), but it did not appear for amorphous PEG (Fig. 4b). The suppressed conformation number of the amorphous state compared with that of liquid PEG provides direct evidence for entropy loss (Fig. 3a).
Comparing PEG/PET with PEG alone, the most significant difference in CH2 vibration modes appeared in the low frequency range of 1300–1200 cm−1. In the crystalline state at 295 K for single PEG (Fig. 4a), the bands at 1282 cm−1 (\({\uptau}_{{\mathrm{CH}}_{2}}\)-1, green track line) and 1241 cm−1 (\({\uptau}_{{\mathrm{CH}}_{2}}\)-2, blue track line) were attributed to the CH2 twisting vibrations (denoted as \({\uptau}_{{\mathrm{CH}}_{2}}\)). After PEG was fixed by PET (Fig. 4b), the corresponding \({\uptau}_{{\mathrm{CH}}_{2}}\) signals at 295 K appeared at a lower frequency of 1280 cm−1 (\({\uptau}_{{\mathrm{CH}}_{2}}\)-1, green track line) and a higher frequency of 1247 cm−1 (\({\uptau}_{{\mathrm{CH}}_{2}}\)-2, blue track line)30,34, indicating a redshift for \({\uptau}_{{\mathrm{CH}}_{2}}\)-1 and a blueshift for \({\uptau}_{{\mathrm{CH}}_{2}}\)-2, respectively. This behavior is quite different from that of the aforementioned \({\upnu}_{{\mathrm{sCH}}_{2}}\), \({\updelta}_{{\mathrm{CH}}_{2}}\) and \({\upomega}_{{\mathrm{CH}}_{2}}\) signals, the wavenumbers of which all remained the same for fixed and free PEG in the 295 K crystalline state (Fig. 4a, b). Moreover, with the SL-PT of single PEG, the bands of \({\uptau}_{{\mathrm{CH}}_{2}}\)-1/2 both showed a sharp blueshift, i.e., from 1282 cm−1 to 1298 cm−1 and from 1241 cm−1 to 1251 cm−1, respectively (Fig. 4a, c). The opposite shifts occurred during the SS-PT of PEG/PET, where the \({\uptau}_{{\mathrm{CH}}_{2}}\)-1 band (1280 cm−1) redshifted to 1274 cm−1 but the \({\uptau}_{{\mathrm{CH}}_{2}}\)-2 band (1247 cm−1) blueshifted to 1257 cm−1 (Fig. 4b, d). For PEG, the \({\uptau}_{{\mathrm{CH}}_{2}}\) band is more closely related to C–C–O skeletons compared to the \({\upnu}_{{\mathrm{sCH}}_{2}}\), \({\updelta}_{{\mathrm{CH}}_{2}}\) and \({\upomega}_{{\mathrm{CH}}_{2}}\) bands35. The highly disordered C–C–O skeleton of liquid PEG aggravates both vibrations of \({\uptau}_{{\mathrm{CH}}_{2}}\)-1 and \({\uptau}_{{\mathrm{CH}}_{2}}\)-2, while possible interchain entanglement between PEG and PET in PEG/PET may make the C–C–O skeleton less disordered and then lead to weakening of the partial \({\uptau}_{{\mathrm{CH}}_{2}}\) vibration. This is also one cause for the entropy loss of amorphous PEG. Moreover, the obviously slower shift of the \({\uptau}_{{\mathrm{CH}}_{2}}\) peaks was seen during the SS-PT compared to that during the SL-PT (Fig. 4c, d), which agrees well with the ΔStr trend of slowing down for PEG/PET compared to that for single PEG (Fig. 3a). Such good consistency evidences the reliability of using IR spectra in the interpretation of the phase transition process. The detailed analysis of the bands at lower frequencies of 1200–1000 cm−1 (red zone, Fig. 4a, b), mainly involving the O–C–C–O skeletons of PEG chains, is given in SI-2.3.
To determine the possible difference in conformation order for the low-temperature crystalline state and high-temperature liquid/amorphous state, DFT calculations on the IR spectra of the PEG molecules with free (denoted as free-PEG) and fixed –OH groups at both ends (denoted as fixed-PEG) were performed. Since the scissoring (\({\updelta}_{{\mathrm{CH}}_{2}}\)), wagging (\({\upomega}_{{\mathrm{CH}}_{2}}\)), and twisting (\({\uptau}_{{\mathrm{CH}}_{2}}\)) vibrations of the CH2 primary group in the PEG chain reflect the main difference before and after PEG solidification (particularly for the high-temperature liquid/amorphous phase, see Fig. 4a, b), we mainly focused on these 3 vibration modes for analysis (Table 1).
For the crystalline state of the PEG molecule, the internal rotation conformations of the O–CH2, CH2–CH2, and CH2–O bonds in the C–C–O skeletons mainly involve TGT and TTT (“T” denotes trans while “G” denotes gauche, see Fig. 5a) according to the literature30,36. Although the calculated wavenumbers of the IR spectra were slightly different from the experimental observations, the evolution trend was entirely the same for fixed-PEG and free-PEG (see details given in SI-3), suggesting that the PEG molecules after fixation by PET still had the two modes of TGT and TTT conformations in the crystalline state.

Conformations of PEG chains in the a crystalline state for free-PEG and fixed-PEG, b liquid state for free-PEG, and c amorphous state for fixed-PEG. d Schematic diagram of the conformational change of the ··O–C–C–O·· skeleton of the PEG chain.
The possible conformations of the high-temperature liquid/amorphous state were further calculated for free-PEG and fixed-PEG. In the O–CH2–CH2–O skeleton chains of the PEG molecule, each pair, i.e., O–CH2, CH2–CH2, and CH2–O, have two possible conformation orders, trans (T) and gauche (G). Once CH2–CH2 is fixed as gauche or trans, the possible conformations are TGT, TGG, GGG or TTT, GTG, TTG (see Fig. 5b).
For the liquid phase of free-PEG, the calculations indicated that the double bands of gauche \({\updelta}_{{\mathrm{CH}}_{2}}\) (1475 and 1462 cm−1) merged into a single band (1471 cm−1), which still corresponds to the gauche conformation between C–C bonds. The wavenumber of 1471 cm−1 appeared for both the calculated TGG and GGG conformations. Moreover, a new trans conformation of \({\updelta}_{{\mathrm{CH}}_{2}}\) (1489 cm−1) appeared for the calculated GTG conformation. Similarly, for the gauche \({\upomega}_{{\mathrm{CH}}_{2}}\) signal, the calculated double bands (1393 and 1379 cm−1) also collapsed into a single band (1392 cm−1) during the SL-PT, which appeared in the calculated TGG. Furthermore, a new trans conformation of \({\upomega}_{{\mathrm{CH}}_{2}}\) (1350 cm−1) also appeared in the calculated GTG conformation. For the \({\uptau}_{{\mathrm{CH}}_{2}}\) band reflecting the degree of disorder of the C–C–O skeleton35,37, the calculated wavenumbers (1312, 1308 cm−1) appeared in each conformation of TGT, TGG, GGG, TTT, GTG, TTG. All these calculated results are in line with the experimentally observed evolution of the gauche conformations of \({\updelta}_{{\mathrm{CH}}_{2}}\) and \({\upomega}_{{\mathrm{CH}}_{2}}\) and the emergence of new trans conformations of \({\updelta}_{{\mathrm{CH}}_{2}}\) and \({\upomega}_{{\mathrm{CH}}_{2}}\) during the SL-PT (Fig. 4a and Table 1). Therefore, it is reasonable to infer that all 6 conformations of TGT, TGG, GGG, TTT, GTG, and TTG coexisted in the high-temperature liquid phase of free-PEG, which is also consistent with previously reported Raman and theoretical results30,34.
For the calculated high-temperature amorphous phase of fixed-PEG by constraining –OH groups at both ends, although the collapse of double bands (1475 and 1462 cm−1) into a single band (1471 cm−1) for gauche \({\updelta}_{{\mathrm{CH}}_{2}}\) was still observed during the crystalline→amorphous SS-PT, the wavenumber 1471 cm−1 only appeared for TGG but disappeared for GGG, implying the possible disappearance of the GGG conformation. Moreover, the new trans conformation of \({\updelta}_{{\mathrm{CH}}_{2}}\) (1489 cm−1) observed for the GTG conformation of free-PEG disappeared for the calculated fixed-PEG, suggesting the disappearance of the GTG conformation for the amorphous solid state of PEG. Analogously, gauche \({\upomega}_{{\mathrm{CH}}_{2}}\) underwent similar evolution during the SS-PT; that is, the double bands (1393 and 1379 cm−1) merged into a single band (1388 cm−1), which still appeared for the calculated TGG conformation. However, the new trans \({\upomega}_{{\mathrm{CH}}_{2}}\) signal (1350 cm−1) observed for the GTG conformation of free-PEG also disappeared, further proving the disappearance of the GTG conformation. Moreover, for \({\uptau}_{{\mathrm{CH}}_{2}}\), the calculated wavenumbers (1312, 1308 cm−1), observed for each of the TGT, TGG, GGG, TTT, GTG, TTG conformations of free-PEG, disappeared for the calculated GGG and GTG conformations, while they only appeared in the TGT, TGG, TTT, TTG conformations for fixed-PEG, further suggesting the disappearance of both the GGG and GTG conformations for the amorphous solid state of PEG (see the diagram shown in Fig. 5c).
The formation of various conformations in the high-temperature liquid/amorphous phase is closely related to the rotations of the C–O bonds in the ··O–C–C–O·· skeletons activated by temperature. The rotation of one C–O bond in ··O–C–C–O·· is enough for the formation of TTG/TGG from TTT/TGT (Fig. 5d). That is, the rotation of any C–O on the left or right hand of the ··O–C–C–O·· skeleton can equally convert TGT into TGG and TTT into TTG. However, simultaneous rotations of at least two C–O bonds in ··O–C–C–O·· are required for the formation of GGG or GTG (Fig. 5d). That is, TTT→GTG/GGG; TGT→GGG/GTG. Such a conversion has a larger resistance and requires higher energy. Therefore, after the –OH groups at both ends of PEG are fixed, the free rotations of the C–O bonds will be limited, making the formation of GGG and GTG conformations more difficult or causing these conformations to even disappear for amorphous PEG.
The DFT calculations reasonably indicated the conformational changes when the liquid PEG was fixed into an amorphous solid by PET. There were 6 conformations of TGT, TGG, GGG, TTT, GTG, TTG coexisting in the liquid free-PEG, while only 4 conformations of TGT, TGG, TTT, TTG coexisted in the amorphous fixed-PEG. The reduction in the conformation order explains the reduced entropy experimentally observed for amorphous PEG. However, since the theoretical statistical weights of the GGG and GTG conformations are small (i.e., GGG: 1/64 and GTG: 1/128)30, the entropy and hence the BCE still remain large for the fixed-PEG (Fig. 3a, Fig. 6).

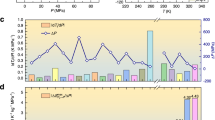
a Heat flow measured at a temperature rate of 1 K/min under selected pressures; the inset shows the transition temperature Tt vs. temperature. b Entropy-temperature (S-T) curves without specific heat; the inset shows the S-T curves including specific heat. c Barocaloric entropy change ΔSp and d adiabatic temperature change ΔTp measured under a pressure of P ≤ 0.1 GPa. e ΔSp and f ΔTp obtained by linearly extrapolating the S curves to P = 0.4 GPa, and accordingly the obtained reversible g ΔSrev and h ΔTrev. The atmospheric pressure and P were taken as the initial values for the compression and decompression processes during the calculations.
To determine the BCE and its reversible features for PEG10000/PET15000, we studied BCE by a quasi-direct method1,9. A commercial high-pressure DSC was employed to measure the variable pressure heat flow curves. The measured heat flow after subtracting the baseline is shown in Fig. 6a. The transition temperature Tt exhibited a high sensitivity to pressure, and Tt increased almost linearly with pressure by a driving rate of approximately dTc/dP = 97 K/GPa (inset of Fig. 6a). Based on the heat flow curves, the calculated entropy without and with including specific heat capacity are shown in Fig. 6b and its insets. Then, the barocaloric entropy change (ΔSp, Fig. 6c) and adiabatic temperature change (ΔTp, Fig. 6d) were obtained by the equations ΔSp (T, P1 → P2) = S (T, P2) − S (T, P1) and ΔTp (S, P1→ P2) = T (S, P2) − T(S, P1), where the atmospheric pressure was taken as the initial and end pressures for the compression and decompression processes, respectively. Accordingly, the maximum ΔSp driven by a low pressure of 0.1 GPa reached 416 J·kg−1·K−1, almost reaching the maximum ΔStr ∼ 426 J·kg−1·K−1 during the SS-PT (Fig. 3a). The achieved ΔSp is larger than that of many other BCE materials (Table 2). Similarly, the obtained ΔTp was 7.3 K under 0.1 GPa.
Due to the pressure limitation of our DSC with an available maximum P = 0.1 GPa, we could not directly obtain the BCE behavior at a higher pressure by the quasi-direct method. Fortunately, Tt shows an almost linear dependence on pressure at P ≤ 0.1 GPa (see the inset of Fig. 6a). If we suppose dT/dP remains constant at a higher pressure P ≤ 0.4 GPa, ΔSp and ΔTp at 0.4 GPa can be accessed by linearly extrapolating the S curves to higher hydrostatic pressure, as shown in Fig. 6e, f. Thus, one can see that a slightly higher driven pressure, i.e., 0.13 GPa, can be enough to complete the phase transition and make the ΔSp reach the maximum, i.e., ΔSp ∼ 426 J·kg−1·K−1.
Subsequently, the reversible isotherm entropy change (ΔSrev) and reversible adiabatic temperature change (ΔTrev) can be straightforward, as shown in Fig. 6g, h. The overlap of ΔSp when applying and removing pressure represents the ΔSrev, while the ΔTrev was similarly obtained excluding the influence of thermal hysteresis14. It is expected that ΔSrev can reach the maximum, i.e., ΔSrev ∼ 426 J·kg−1·K−1, while ΔTrev ∼ 12.6 K at 0.4 GPa. Such performance of BCE is indubitably attractive compared to that of most other BCE materials (Table 2). Note that there is almost no reversible entropy change under a small pressure, even 0.4 GPa, for NPG ((CH3)2C(CH2OH)2), AMP ((NH2)(CH3)C(CH2OH)2), and TRIS ((NH2)C(CH2OH)3). The giant BCE of PEG/PET originates from the large latent heat of the SS-PT owing to the remaining large entropy in the amorphous state and the high-pressure sensitivity of Tt (dTc/dP = 97 K/GPa).
Conclusions
In conclusion, a new type of full solid-state colossal barocaloric material around room temperature was fabricated by exploiting the amorphous high entropy inherited from huge disorder of liquid phase in solidified PEG10000 by introducing 5 wt.% PET15000 for copolymerization. The formed hydrogen bonds between the terminated –OH of PEG and the rigid PET chains solidified the flow of PEG segments. As a result, liquid PEG converted into an amorphous solid, while colossal latent heat retains. TEM combined with XRD measurements demonstrated the amorphous nature of the high-temperature phase after fixation by PET. The SS-PT showed a high sensitivity to pressure, and hence colossal ΔSp of ∼416 J·kg−1·K−1 under a low pressure of 0.1 GPa was measured, which exceeds the performance of most other BCE materials.
To disclose the origin of the high remaining entropy of the amorphous solid phase, IR measurements and DFT calculations were performed. The conformation orders remain the same for the crystalline states of free- and fixed-PEG. For the PEG amorphous phase, compared to its liquid phase, the experimental disappearances of wagging (\({\upomega}_{{\mathrm{CH}}_{2}}\)) vibrations at ~1324 cm−1 and scissoring (\({\updelta}_{{\mathrm{CH}}_{2}}\)) vibrations at ~1485 cm−1 suggested the conformational reduction of the PEG chain. DFT calculations gave the specific conformational changes from a liquid to an amorphous solid state. The disappearance of the GGG and GTG conformations, which account for small statistical weights, explained the reduced but still large entropy change observed during the SS-PT. The large remaining entropy of the amorphous solid and its high sensitivity to pressure make PEG/PET an attractive potential full solid-state refrigerant for refrigeration applications. Our work provides a new strategy for exploring novel solid-state barocaloric materials by utilizing the high entropy inherited from huge disordering of liquid phases in various SL-PT materials.
References
Mañosa, L. & Planes, A. Materials with giant mechanocaloric effects: cooling by strength. Adv. Mater. 29, 1603607 (2017).
Rudolph, K., Pathak, A. K., Mudryk, Y. & Pecharsky, V. K. Magnetostructural phase transitions and magnetocaloric effect in (Gd5-xScx)Si1.8Ge2.2. Acta Mater. 145, 369–376 (2018).
Franco, V. et al. Magnetocaloric effect: from materials research to refrigeration devices. Prog. Mater. Sci. 93, 112–232 (2018).
Tušek, J. et al. The elastocaloric effect: a way to cool efficiently. Adv. Energy Mater. 5, 1500361 (2015).
Cazorla, C. Novel mechanocaloric materials for solid-state cooling applications. Appl. Phys. Rev. 6, 041316 (2019).
Aznar, A. et al. Giant and reversible inverse barocaloric effects near room temperature in ferromagnetic MnCoGeB0.03. Adv. Mater. 31, 1903577 (2019).
Matsunami, D., Fujita, A., Takenaka, K. & Kano, M. Giant barocaloric effect enhanced by the frustration of the antiferromagnetic phase in Mn3GaN. Nat. Mater. 14, 73–78 (2015).
Lloveras, P. et al. Giant barocaloric effects at low pressure in ferrielectric ammonium sulphate. Nat. Commun. 6, 8801 (2015).
Moya, X., Kar-Narayan, S. & Mathur, N. D. Caloric materials near ferroic phase transitions. Nat. Mater. 13, 439–450 (2014).
Fähler, S. & Pecharsky, V. K. Caloric effects in ferroic materials. MRS Bull. 43, 264–268 (2018).
Romanini, M. et al. Giant and reversible barocaloric effect in trinuclear spin-crossover complex Fe3(bntrz)6(tcnset)6. Adv. Mater. 33, 2008076 (2021).
Vallone, S. P. et al. Giant barocaloric effect at the spin crossover transition of a molecular crystal. Adv. Mater. 31, 1807334 (2019).
Li, B. et al. Colossal barocaloric effects in plastic crystals. Nature 567, 506–510 (2019).
Aznar, A. et al. Reversible and irreversible colossal barocaloric effects in plastic crystals. J. Mater. Chem. A 8, 639–647 (2020).
Lloveras, P. et al. Colossal barocaloric effects near room temperature in plastic crystals of neopentylglycol. Nat. Commun. 10, 1803 (2019).
Li, F. B. et al. Understanding colossal barocaloric effects in plastic crystals. Nat. Commun. 11, 4190 (2020).
Salyan, S., Suresh, S. & Sarath Reddy, A. Low melt alloy enhanced solid-liquid phase change organic sugar alcohol for solar thermal energy storage. J. Mol. Liq. 266, 29–42 (2018).
Lin, J. et al. Colossal and reversible barocaloric effect in liquid-solid-transition materials n-alkanes. Nat. Commun. 13, 596 (2022).
Tang, Y. R., Gao, D. L., Guo, Y. F., Wang, S. Q. & Deng, T. L. Supercooling and phase separation of inorganic salt hydrates as PCMs. Appl. Mech. Mater. 71-78, 2602–2605 (2011).
Purohit, B. K. & Sistla, V. S. LAPONITE® based hydrogel for cold thermal energy storage application. Bull. Mater. Sci. 44, 110 (2021).
Ansu, A. K. et al. A cycling study for reliability, chemical stability and thermal durability of polyethylene glycols of molecular weight 2000 and 10000 as organic latent heat thermal energy storage materials. Int. J. Energy Res. 44, 2183–2195 (2020).
Pielichowska, K. & Pielichowski, K. Phase change materials for thermal energy storage. Prog. Mater. Sci. 65, 67–123 (2014).
Kim, H. J., Reddi, Y., Cramer, C. J., Hillmyer, M. A. & Ellison, C. J. Readily degradable aromatic polyesters from salicylic acid. ACS Macro Lett. 9, 96–102 (2020).
Bourland, L. G. Polyethylene terephthalate graft copolymers acting as an interfacial modifier in rubber modified polyethylene terephthalate compounds. J. Plast. Film. Sheet 31, 363–378 (2015).
Chen, S. et al. General decomposition pathway of organic-inorganic hybrid perovskites through an intermediate superstructure and its suppression mechanism. Adv. Mater. 32, 2001107 (2020).
Huang, G. M. et al. In situ TEM investigation of electron beam-induced ultrafast chemical lithiation for charging. J. Mater. Chem. A 8, 648–655 (2020).
Wei, B. et al. In situ TEM investigation of electron irradiation induced metastable states in lithium-ion battery cathodes: Li2FeSiO4 versus LiFePO4. ACS Appl. Energy Mater. 1, 3180–3189 (2018).
Liang, X. H., Guo, Y. Q., Gu, L. Z. & Ding, E. Y. Crystalline-amorphous phase transition of a poly(ethylene glycol)/cellulose blend. Macromolecules 28, 6551–6555 (1995).
Alkan, C., Günther, E., Hiebler, S., Ensari, Ö. F. & Kahraman, D. Polyethylene glycol-sugar composites as shape stabilized phase change materials for thermal energy storage. Polym. Compos. 33, 1728–1736 (2012).
Matsuura, H. & Miyazawa, T. Vibrational analysis of molten poly(ethylene glycol). J. Polym. Sci. PART A-2 7, 1735–1744 (1969).
Bailey, F. E. & Koleske, J. V. Poly(ethylene oxide). (Academic Press, Inc., 1976).
Pielichowska, K. et al. PEO/fatty acid blends for thermal energy storage materials. Structural/morphological features and hydrogen interactions. Eur. Polym. J. 44, 3344–3360 (2008).
Yoshihara, T., Tadokoro, H. & Murahashi, S. Normal vibrations of the polymer molecules of helical conformation. IV. Polyethylene oxide and polyethylene‐d4 oxide. J. Chem. Phys. 41, 2902–2911 (1964).
Koenig, J. L. & Angood, A. C. Raman spectra of poly(ethy1ene glycols) in solution. J. Polym. Sci. PART A-2 8, 1787–1796 (1970).
Davison, W. H. T. Infrared spectra and crystallinity. Part III. Poly(ethylene glycol). J. Chem. Soc. 2431, 3270–3274 (1955).
Miyazawa, T., Fukushima, K. & Ideguchi, Y. Molecular vibrations and structure of high polymers. III. Polarized infrared spectra, normal vibrations, and helical conformation of polyethylene glycol. J. Chem. Phys. 37, 2764–2776 (1962).
Han, L., Peng, L., Cai, L. Y., Zheng, X. M. & Zhang, F. S. CH2 scissor and twist vibrations of liquid polyethylene glycol-Raman spectra and density functional theory calculations. Acta Phys. Chim. Sin. 33, 1043–1050 (2017).
Aznar, A. et al. Giant barocaloric effects over a wide temperature range in superionic conductor AgI. Nat. Commun. 8, 1851 (2017).
Min, J., Sagotra, A. K. & Cazorla, C. Large barocaloric effects in thermoelectric superionic materials. Phys. Rev. Mater. 4, 015403 (2020).
Aznar, A. et al. Reversible colossal barocaloric effects near room temperature in 1-X-adamantane (X = Cl, Br) plastic crystals. Appl. Mater. Today 23, 101023 (2021).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFB3501202, 2020YFA0711502, 2019YFA0704900, 2022YFB3505201, 2021YFA1400300, 2022YFA1403302, 2018YFA0305704), the National Natural Sciences Foundation of China (52088101, U1832219, 51971240, 52101228, 11921004), the Strategic Priority Research Program (B, XDB33030200) and the Key Research Program (ZDRW-CN-2021-3) of the Chinese Academy of Sciences (CAS). We thank Yongzhao Zhang for help with the TEM measurements.
Author information
Authors and Affiliations
Contributions
F.H. and B.S. conceived the study. Z.B.Y., J.H., and Y.G. prepared the samples. Y.W., B.W., and Z.T. helped to prepare samples. Z.B.Y. and H.Z. carried out XRD and TEM measurements and corresponding analyses. Y.L., C.Z., and Z.Y. helped with the TEM analysis. Z.B.Y., C.L., S.Y., and Y.L. performed the IR measurements and analysis. Z.B.Y., D.W., and Y.L. performed the DFT calculations and analysis. Z.B.Y. performed the DSC measurements and barocaloric analysis. J.W., Y.C., J.S., and T.Z. participated in the data analysis. F.H., Z.B.Y., H.Z., and J.W. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, Z., Zhou, H., Hu, F. et al. Colossal barocaloric effect achieved by exploiting the amorphous high entropy of solidified polyethylene glycol. NPG Asia Mater 14, 96 (2022). https://doi.org/10.1038/s41427-022-00448-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-022-00448-7
This article is cited by
-
Low pressure reversibly driving colossal barocaloric effect in two-dimensional vdW alkylammonium halides
Nature Communications (2024)
