
Abstract
The study is to explore the role of ferroptosis-related genes (FRGs) in the occurrence and development of gastric cancer (GC), and to construct a new prognosis signature to predict the prognosis in GC. Clinical information and corresponding RNA data of GC patients were downloaded from TCGA and GEO databases. Consensus clustering was performed to identify new molecular subgroups. ESTIMATE, CIBERSORT, McpCounter and TIMER algorithm were used to analyze the infiltration of immune cells in two molecular subgroups. LASSO algorithm and multivariate Cox analysis were used to construct a prognostic risk signature. Functional analysis was conducted to elucidate the underlying mechanisms. Finally, the FRPGs were verified by Quantitative Real-Time PCR. We obtained 16 FRGs and divided GC patients into two subgroups by consistent clustering. Cluster C1 with a higher abundance of immune cell infiltration but lower probability in response to immunotherapy, it was reasonable to speculate that Cluster C1 was in accordance with the immune rejection type. Functional analysis showed that the biological process of DEGs in training cohort mainly included immune globulin, and human immune response mediated by circulating immune globulin. GSEA analysis showed that compared with Cluster C2, Cluster C1 showed lower expression in lipid metabolism. The nomogram combined with risk signature and clinical features can accurately predict the prognosis of GC patients. We identified two molecular subtypes, Clusters C1 and C2. In Cluster C1, patients with poor prognosis present with a hyperimmune status and low lipid metabolism, and we speculate that Cluster C1 was in accordance with the immune rejection type. The risk model based on FRPGs can accurately predict the prognosis of GC. These results indicated that ferroptosis is associated with TIME, and deserved considerable attention in determining immunotherapy treatment strategy for GC patients.
Similar content being viewed by others


Introduction
A preprint has previously been published1.
Gastric cancer (GC) is the fourth leading cause of cancer-related death2. The overall survival of patients with GC varies widely in different regions of the world. For example, the 5-year survival rate is 31% in the United States, 19% in the United Kingdom, and 26% in Europe3. Because the early stage of GC is usually asymptomatic, it is mostly late when it is discovered, resulting in a 5-year overall survival rate (OS) of less than 40%4. Therefore, exploring new and more effective treatment methods has become a problem to be solved.
The occurrence and development of tumors are closely related to cell death. Ferroptosis is a form of non-apoptotic cell death found in recent years5. Triggered by lipid reactive oxygen species (ROS)6. Ferroptosis is related to the occurrence of many kinds of tumors, including lung cancer7, breast cancer8, colorectal cancer9, and GC10. In recent years, prognosis models of ferroptosis associated with various diseases have been constructed, such as adrenocortical carcinoma11, and pancreatic cancer12. Many ferroptosis genes including GPX4, SLC7A11 and NRF2 have been found to be promising targets for inducing tumor cell death. For example, an innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment13. SLC7A11 promoted the proliferation, migration, and invasion of renal cancer cells by enhancing GPX4 output, which in turn inhibits ferroptosis14. Therefore, targeting ferroptosis may be a new strategy for cancer treatment.
Tumor immunotherapy, as a new treatment method based on human immune system, plays an anti-tumor role by immune regulation. The use of checkpoint inhibitors has been proved to be of great significance in improving the objective remission rate of tumors and prolonging the survival time of patients15,16. In the last decade, molecular subtype-based classification of GC offers opportunities for personalized treatment. Biomarkers, particularly microsatellite instability (MSI), programmed cell death ligand 1 (PD-L1), human epidermal growth factor receptor 2 (HER2), tumor mutational burden, and Epstein-Barr virus. The use of for first-line immunotherapy for advanced GC, HER2- patients can choose Nivolumab or Sintilimab combined with chemotherapy, and HER2 + patients are recommended to be treated with Trastuzumab combined with Pembrolizumab combined with chemotherapy. In addition to immune checkpoint inhibitors, cellular immunotherapy may become another effective weapon for the treatment of advanced GC17. Tumor infiltrating immune cells (TIIC) are related to many kinds of tumor prognosis and immunotherapy response18,19,20. For example, M2 macrophages are enriched in bladder cancer tissue, which can be used as a potential immunotherapy target for bladder cancer21. Many kinds of tumor immunotherapy related to natural killer cells (NK) have also entered the clinical trial stage22. It has been proved that TIME is closely related to the pathogenesis of GC23. Therefore, mining immune-related ferroptosis targets is an effective way to optimize tumor immunotherapy24.
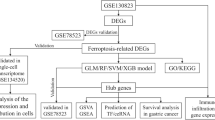
In this study, we comprehensively analyzed the FRGs to explore the influence of ferroptosis on the TIME and survival of patients with GC. In addition, we constructed a FRGs-risk signature to evaluate the prognostic value in GC, which will provide a new strategy for targeted and individualized treatment of GC. The protocol of our study procedures is shown in Fig. 1.

The protocol of our study procedures.
Method
Data collection and analysis
The FPKAPLAN-MEIER ANALYSIS gene expression profile of GC (TCGA-STAD) was obtained from TCGA database (https://portal.gdc.cancer.gov/). As a training cohort, we obtained 443 clinical samples, including 375 samples of cancer tissues and 68 samples of adjacent tissues. We extracted the mapping information of GeneSymbol and ENSG_ID, mapped the ENSG_ID to GeneSymbol, and when there were multiple matches, took the median, and finally obtained the transformed expression spectrum. Finally, the data was further standardized by log2(X + 1) transformation and we used samples with complete clinical information for subsequent analysis. The microarray data GSE84426 and GSE84437 were downloaded from GEO database (http://www.ncbi.nlm.http://nih.gov/geo/) through GPL6947 application platform. As validation cohorts, we obtained 509 GC tissue samples. Clinical information of the above patient is shown in Table 1. 259 genes related to ferroptosis were collected from FerrDB (http://www.zhounan.org/ferrdb/) database25, including driver, suppressor, and marker, Species was defined as Human.
Identification of molecular subtype
The genes related to the prognosis of GC were screened out by univariate Cox analysis. We intersected the prognosis-related genes of GC with ferroptosis genes by Venn diagram to obtain ferroptosis-related prognosis genes (FRPGs). Cluster analysis was performed using ConsensusClusterPlus, using agglomerative pam clustering with a 1-pearson correlation distances and resampling 80% of the samples for 10 repetitions. The optimal number of clusters was determined using the empirical cumulative distribution function plot.
Immune analysis
Immunological analysis was used to explore the immune differences between the two subgroups. Estimate algorithm (estimation of violent and immune cells in malignant tumor organization using expression) is used to evaluate the proportion of immune-matrix components in TIME, include Stromal Score (reflecting the presence of matrix), Immune Score (reflecting the level of immune cell infiltration) and ESTIMAT Score (comprehensive score of immunity and matrix)26. The higher the corresponding score, the larger the proportion of corresponding components in TIME. Using MCP Counter and TIMER databases27 to calculate the abundance of immune infiltrating cells. CIBERSORT algorithm was used to estimate the data of tumor infiltrating immune cells 28.
Establishment of prognostic risk signature based on FRPGs
The “glmnet” R package29 was used for LASSO analysis to further select hub prognostic markers, and the minimum lambda was defined as the optimal value. According to the risk score of the prognosis signature, GC patients were divided into high and low risk groups. Kaplan–Meier survival curve and time-dependent ROC curve were used to analyze and compare the survival situation between the two groups. Immunohistochemical (IHC) staining of hub FRPGs were examined by human protein atlas (HPA) (https://www.proteinatlas.org/about/download). Maftools was used to calculate the mutation of hub FRPGs, and “ggplot2” package30 was used to draw the mutation distribution map.
Validation of prognostic risk signature
Multivariate Cox analysis and subgroup analysis were used to evaluate the independence of the prognosis signature and the clinical characteristics (including Gender, Age,Grade, and Stage) of patients. The GEO validation cohort was used to verify the accuracy of the established prognosis signature. Combining the prognosis signature and clinical features, nomogram was constructed to predict the 1,3 and 5-year survival rate of GC patients.
Functional enrichment analysis
Differentially expressed genes (DEGs) between the two clusters were identified using R package ‘‘Limma’’31 in the training cohort, the false discovery rate (FDR) was less than 0.05, and the difference multiple was 1.5 times as the screening standard. GSEA enrichment analysis was carried out according to DEGs. Metascape software was used to construct PPI network of DEGs32. Moreover, we also performed survival analysis on samples in the GEO validation cohorts, divided the samples into high and low risk groups according to risk scores, and screened out DEGs by “limma” R package31. Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis33,34 were performed to enrich associated pathways.
Cell culture
GC cell lines HGC-27 and normal human gastric epithelial cell lines GSE-1 were purchased from Nanjing KGI Biotechnology Company. All cells were cultured in RPMI-1640 medium supplemented with 5% fetal bovine serum at 37 °C in a humidified atmosphere with 5% CO2.
Quantitative real-time PCR
Total RNA was extracted from cells using the TRIzol kit according to the manufacturer's protocol. Reverse transcription was performed using the PrimeScript RT kit (Takara, China) according to the manufacturer's instructions. SYBR PrimeScript RT-PCR Kit (Takara) is used for quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis. The 2−ΔΔCt statistic was used to calculate the expression level of the gene. The specific sequences of the different primers used in this study are included in Supplementary Table S1.
Statistical analysis
All statistical analysis in this paper was carried out by R software and GraphPad Prism. Student’s t-test was used for statistical analysis between two groups, and one-way ANOVA analysis was selected flexibly when there were three or more groups. P < 0.05 was considered to be statistically significant.
Statement
All methods were carried out in accordance with relevant guidelines and regulations. Ethical approval is not applicable for this study.
Results
Identification of molecular subtypes based on FRGs
Through univariate Cox analysis, we obtained 2,381 GC prognosis-related genes, and 16 FRPGs were obtained by intersecting 2,381 GC prognosis-related genes with 259 ferroptosis genes (Fig. 2A). The consensus clustering approach was conducted to divide the GC patients in the training cohort. The optimal clustering stability was identified when K = 2 (Fig. 2B–E). Cluster C1 included 210 patients, while Cluster C2 included 197 patients. The two subgroups were visualized by heatmap (Fig. 2F). Kaplan–Meier survival curves of two different subgroups showed that Cluster C2 showed significant median survival advantage, while Cluster C1 showed poor prognosis (Fig. 2G).

Molecular subtype identification based on FRGs. (A) Venn diagram, a prognostic gene for GC associated with ferroptosis. The pink represents GC-related genes, blue represents ferroptosis-related genes. (B–E) K = 2 was identified the optimal value for consensus clustering. (B) Represents Cumulative distribution curve. (C) Represents Area under distribution curve. (D) represents Heatmap of consensus clustering. (E) Represents Sample clustering consistency. (F) Heatmap of FRPGs in two subgroups. (G) Kaplan–Meier survival curves for two subgroups.
Different TIME and immune status in the two molecular subtypes
The ESTIMATE algorithm showed significantly higher ESTIMATE scores (P < 0.0001), higher Immune score (P < 0.01), and higher Stromal score (P < 0.0001) in Cluster C1 compared with Cluster C2 (Fig. 3A). The MCPCounter algorithm showed that the expression levels of B-lineage (P < 0.00), Myoid-Dendritic-cells (P < 0.0001), Neutrophils (P < 0.01), Endothelial-cells (P < 0.0001) and Fibroblasts (P < 0.0001) in Cluster C1 were significantly higher than those in Cluster C2 (Fig. 3B). The TIMER algorithm showed that fibrolasts (p = 7.80e−12), CD4 + T cells (p = 0.0009), and B cells (p = 0.0023) were significantly higher in Cluster C1 than in Cluster C2. Macrophases-M2 and Tregs in two Cluster was no significant difference (Fig. 3C,D). The CIBERSORT algorithm indicated Macrophases-M0 (p = 0.0000052), CD4 Memory Activated-T cells (p = 0.00024), Macrophases-M1 (p = 0.001), Nave-B-cells (p = 0.001), NK cells (P = 0.02) were significantly higher in Cluster C1 than in Cluster C2. Macrophases-M2 in two Cluster was no significant difference (Fig. 3E,F), suggesting a relatively low immune status in Cluster C2. These results demonstrated that the TIME and immune status of the two molecular subtypes differed significantly. Cluster C1 with a poor prognosis had a high immune status, so we speculate that Cluster C1 is immune rejection type.

Immune analysis of two molecular subtypes. (A) Stromal score, Immune score, ESTIMATE score and calculated by ESTIMATE algorithm (*p < 0.05; **p < 0.01; ***p < 0.001). (B) Abundance of ten immune filtrating cells evaluated by MCPcounter algorithm (*p < 0.05; **p < 0.01; ***p < 0.001). (C) Heatmap depicting the enriching level of immune related cells evaluated by TIMER. (D) Statistical analysis of immune related cells evaluated by TIMER (*p < 0.05; **p < 0.01; ***p < 0.001). (E) Heatmap depicting the enriching level of 25 immune related cells evaluated by CIBERSORT algorithm. (F) Statistical analysis of 25 immune related cells evaluated by CIBERSORT algorithm (*p < 0.05; **p < 0.01; ***p < 0.001).
Establishment of prognostic risk signature based on FRPGs
We signatureed the risk based on LASSO analysis, and we set the lambda value to be 0.029411686746793, Built signature RiskScore = [0.17987688835102 * ZFP36] − [0.2111220517453 * TUBE1] − [0.1164248611519 * SLC1A4] + [1.958393356095 * NOX5] + [0.480740705518259*NOX4] − [0.39552084635819 * NFE2L2] + [2.63582963497227 * MIR9] − [0.0833648850166638*GCH1] + [0.702888544996765 * GABARAPL2] − [0.117744902736126 * CHAC1] + [0.010366289402305 * CAPG] − [0.25896183370357 * ACSL4] − [0.033599913666074 * ACO1], thirteen genes were finally obtained, and three of them were risk genes with the risk ratio greater than 1 (Fig. 4A). The established risk signature successfully classified the GC patients into high risk and low risk groups (Fig. 4B). Kaplan–Meier analysis showed that patients in the low risk group had a better overall survival than those in the high risk group (Fig. 4C). Based on the information such as the survival status and survival time of the patients, we drew the forest map for multivariate survival analysis of FRPGs (Fig. 4D). ROC analysis showed that the risk signature constructed exhibited accurate prediction ability within 5 years, with AUC of 0.69, 0.80 and 0.81 for 1, 3 and 5 years (Fig. 4E). Finally, the TIME of the two groups was evaluated using the ESTIMATE algorithm, and the results showed that the high risk group had a higher ESTIMATE score (P = 1.2e−4), higher Immune score (P = 4.8e−7), and higher Stromal score (P = 5.1e−7) (Fig. 4F). The TIMER database was used to predict the relationship between the FRPGs and the infiltration level of immune cells, and it was found that the FRPGs were closely related to the infiltration of macrophages, B cells, T cells, dendritic cells, and neutrophils (Fig. 4G). The CIBERSORT algorithm indicated that Macrophases-M1 (p < 0.01) and Macrophases-M2 (p < 0.001) were significantly higher in high risk groups than in low risk groups, while Dendritic-cells-activated was significantly higher in low risk groups than in high risk groups (Fig. 4H), suggesting the TIME and immune status of the two groups differed significantly. These results indicated that the risk signature constructed had a strong potential for prognosis prediction of GC patients, and it was significantly correlated with TIME in GC. The HPA database examined the immunohistochemical (IHC) staining of FRPGs (Fig. 5A–J), and found that the protein expressions of ZFP36, TUBE1, NFE2L2, GCH1, GABARAPL2, CHAC1, CAPG, ACSL4, ACO1, and SLC1A4 in GC and normal tissues were significantly different, there was no protein expression of NOX5, MIR9-3, and NOX4 in HPA. In addition, we observed mutations of FRPGs in the training cohort and found that TUBE1 was a dominant gene and therefore better targeted (Fig. 6).



Establishment of prognostic risk signature based on FRPGs in the training cohort. (A) LASSO analysis with suitable lambda. (B) Distribution of survival status, risk score, and heatmap of GC patients in the high and low risk groups. (C) Survival curve of the GC patients in the two groups. (D) Forest map of multi-factor survival analysis. (E) Time-dependent ROC curve of the risk signature. (F) Stromal score, Immune score, and ESTIMATE score in the high and low risk groups (*p < 0.05; **p < 0.01; ***p < 0.001). (G) Correlation between the FRPGs and the infiltration level of immune cells in TIMER database. (H) Statistical analysis of immune related cells evaluated by CIBERSORT algorithm in the two groups (*p < 0.05; **p < 0.01; ***p < 0.001).

Immunohistochemical staining of FRPGs in HPA. The expression of (A) ZFP36, (B) TUBE1, (C) NFE2L2, (D) GCH1, (E) GABARAPL2, (F) CHAC1, (G) CAPG, (H) ACSL4, (I) ACO1, and (J) SLC1A4 in the HPA.

Mutations of FRPGs in the training cohort. The green represents Missense Mutation, blue represents Frame Shift Del, red represents Nonsense Mutation, and orange represents Splice Site.
Validation of prognostic risk signature
We assessed the differences in risk scores among the subgroups by age (Fig. 7A), gender (Fig. 7B), T (Fig. 7C), N (Fig. 7D), Stage (Fig. 7E), and Grade (Fig. 7F) and found no significant differences between the subgroups, indicating that the risk scores were not correlated with the clinical characteristics of the patient. Besides, when the patients were regrouped according to age (Fig. 7G,H), and gender (Fig. 7I,J), the risk signature still exhibited potent predictive performance and those patients with lower risk score enjoyed better prognosis. This indicated that the prognosis signature we constructed can be used to independently predict the prognosis of GC patients.

Association of risk score and clinical characteristics. Differences in risk scores among (A) Age, (B) Gender, (C) T, (D) N, (E) Stage, and (F) Grade. (G,H) Survival curve of GC patients regrouped according to age. (I,J) Survival curve of GC patients regrouped according to gender.
To verify the stability of the prognostic signature developed in the training cohort, we calculated the risk score for each patient in the validation cohort using the same formula as in the build training cohort. Patients in the validation cohort were grouped into high (n = 216) and low risk (n = 293) groups (Fig. 8A), and ROC analysis revealed AUC values of 0.63, 0.65, and 0.69 at 1, 3, and 5 years, respectively (Fig. 8B). The Survival curve showed that the low risk group had a better prognosis than the high risk group (Fig. 8C). In addition, the ESTIMATE algorithm was performed on high risk and low risk group in the validation cohort, and the results showed that the high risk group had higher Stromal score (p < 0.001), higher ESTIMATE score (p < 0.001), and lower Tumor Purity (p < 0.001) (Fig. 8D–G). These results demonstrated that the established risk signature was correlated with TIME and prognosis in GC in the validation cohort.

Validation of prognostic risk signature in validation cohort. (A) Distribution of survival status and risk score in the high and low risk groups. (B) ROC curve of the risk signature in validation cohort. (C) Survival curve of the patients in the high and low risk groups. (D–G) Stromal score, Immune score, Tumor Purity, and ESTIMATE score calculated by ESTIMATE algorithm (*p < 0.05; **p < 0.01; ***p < 0.001). (H) Nomogram integrating risk score and clinical features. (I) Calibration of the nomogram at 1,3, and 5 years in the training cohort (*p < 0.05; **p < 0.01; ***p < 0.001). (J) Calibration of the nomogram at 1,3, and 5 years in the validation cohort (*p < 0.05; **p < 0.01; ***p < 0.001).
Finally, a nomogram integrating the risk signature and clinical features were constructed to predict the prognosis of the GC patients more precisely (Fig. 8H). The 3 years survival rates observed with the nomogram were matched well to the actual survival rates (Fig. 8I), and similar result was also observed in the validation cohort (Fig. 8J), demonstrating that the nomogram could predict the prognosis of GC patients accurately.
Functional enrichment analysis
A total of 570 DEGs were detected compared to Cluster C2, of which 465 genes were up regulated and 105 genes were down regulated in Cluster C1 (Fig. 9A,B). GO enrichment analysis showed that the biological process (BP) of DEGs mainly included cell migration, immune globulin, human immune response mediated by circulating immune globulin. Cellular component (CC) was mainly enriched in the extracellular matrix, and extracellular region part. Molecular function (MF) mainly included immunoglobulin receptor binding, fibronectin binding, and growth factor binding (Fig. 9C–G). The KEGG enrichment analysis showed that DEGs was mainly enriched in the Cell cycle, p53 signaling pathway, IL-17 signaling pathway, MAPK signaling pathway, and PI3K-Akt signaling pathway (Fig. 9H–I). To further explore the relationship between enrichment pathways and prognosis of GC patients, we performed GSEA analysis, and the results showed that compared with Cluster C2, Cluster C1 showed lower expression in lipid metabolism and glutathione metabolism, which might be related to the poor prognosis of GC patients (Fig. 9J). All these results demonstrated that expression of FRPGs were correlated with immunity and ferroptosis, which may be involved in the poor prognosis of GC patients.


Identification of DEGs and Functional Enrichment Analysis. (A) Volcanic map of DEGs in training cohort. (B) Heatmap of DEGs in training cohort. (C–F) BP, CC, MF, and the whole gene ontology (GO) Analysis of DEGs in training cohort. (G) PPI analysis of GO Analysis in training cohort. (H) Circle diagram of KEGG in training cohort. (I) PPI analysis of KEGG enrichment analysis. (J) Heatmap of GSEA analysis results.
Compared with the DEGs in training cohort, a total of 71 DEGs were detected, of which 7 genes were up regulated and 64 genes were down regulated in validation cohort (Fig. 10A,B). KEGG enrichment analysis showed that DEGs was mainly enriched in the Cell cycle, p53 signaling pathway, and MAPK signaling pathway, which consistent with the results in training cohort. In addition, DEGs in validation cohort was also enriched in the Jak-STAT signaling pathway, TNF signaling pathway (Fig. 10C–F).

Identification of DEGs and functional enrichment analysis in GEO cohorts (www.kegg.jp/kegg/kegg1.html). (A) The volcanic map of DEGs. (B) The heatmap of DEGs. (C–F) The BP, CC, MF, and the whole GO of DEGs.
Quantitative Real-Time PCR
We found that the protein expressions of ZFP36, TUBE1, NFE2L2, GCH1, GABARAPL2, CHAC1, CAPG, ACSL4, ACO1, and SLC1A4 in GC and normal tissues were significantly different. Their expression levels were evaluated in GES-1 and HGC-27 by qRT-PCR. Consistently, compared with GES-1, GCH1 (p < 0.01), CAPG (p < 0.05), and TUBE1 (p < 0.05) were significantly upregulated in HGC-27, while ZFP36 (p < 0.05), GABARAPL2 (p < 0.05), NFE2L2 (p < 0.01), and ACSL4 (p < 0.05) were downregulated, but CHAC1, ACO1, and SLC1A4 were no significant difference (Fig. 11A–J). In summary, the potential roles of the FRPGs could also be verified in cell line experiment.

mRNA expression level of FRPGs. mRNA expression level of (A) GCH1, (B) CHAC1, (C) CAPG, (D) TUBE1, (E) ZFP36, (F) GABARAPL2, (G) NFE2L2, (H) ACSL4, (I) ACO1, and (J) SLC1A4 by qRT-PCR (*p < 0.05; **p < 0.01; ***p < 0.001).
Discussion
In recent years, various prognostic signatures have been proposed to predict the prognosis and immune infiltration of malignant tumors35. Including predicting the immune status and prognosis of malignant tumors by screening immune-related genes or FRGs. Few studies have elucidated the effects of the FRGs on TIME and prognosis. Therefore, in order to further verify the effect of ferroptosis on TIME in GC and explore the prognostic value of FRPGs in patients with GC, we constructed a prognostic risk signature based on FRPGs and validated it in the validation cohort.
Firstly, we divided GC patients into two subgroups by consistent clustering. Cluster C2 showed a significant median survival advantage, while Cluster C1 showed a poor prognosis. Subsequently, we performed an immune analysis to explore the role of ferroptosis in TIME. TIME plays a vital role in the prognosis of patients because tumor progression is associated with changes in the surrounding matrix, of which immune cells are a key component36. Therefore, we applied ESTIMATE, MCPCounter, TIMER, and CIBERSORT algorithms to determinate the TIME of the two subgroups. Compared with Cluster C1, GC patients in Cluster C1 had significantly higher ESTIMATE scores, higher Immune score, and higher Stromal score. Our results indicated that Macrophases-M1, CD4 Memory Activated-T cells, NK cells, fibrolasts, CD4 + T cells, B cells were significantly higher in Cluster C1 than in Cluster C2. Macrophases-M2 and Tregs in two Cluster was no significant difference, suggesting a relatively high immune status in Cluster C1. M1 macrophages are linked to antitumor activity, whereas M2 macrophages are associated with cancer progression and metastasis37. These results demonstrated that the TIME and immune status of the two molecular subtypes differed significantly. TIME is generally divided into three categories: immune inflammation, immune rejection and immune desert38. In this study, according to the exhibitions that the TIME of the patients in the Cluster C1 with a higher abundance of immune cell infiltration but lower probability in response to immunotherapy, it was reasonable to speculate that Cluster C1 was in accordance with the immune rejection type. As a successful immunotherapy depends on the ability of innate and adaptive immune cells to penetrate into the tumor parenchyma and eradicate cancer cells. In addition, we also found that the high infiltration of immune cells was accompanied by the activation of the stroma, which could exclude the entry of CD8 + T cells from the tumor parenchyma to the peritumoral stroma rich in fibroblasts and collagen39. We speculated that the activation of the stroma might be one of the reasons for the poor prognosis of Cluster C1, which had high infiltration of immune cells. Therefore, the prognosis of high risk group was often poor.
In order to further verify the effect of ferroptosis on TIME in GC and explore the prognostic value of FRPGs in patients with GC, we constructed a prognostic risk signature based on FRPGs and validated it in the validation cohort. The genes used to establish the risk signature in this study have been shown to be closely related to tumor development and progression. A study had shown that autophagy promotes ferroptosis by degrading anti-ferroptosis factors40, and ZFP36 was a key protein for autophagy and considered to be related to ferroptosis41. NFE2L2, a known transcription factor involved in the encoding of GC development, is overexpressed as a prognostic marker of GC42. OS rate in GC patients with NRF2 positive expression was significantly reduced43. The experiment conducted by Wei44 proved that GCH1 induced immunosuppression through a 5-HTP-AHR-ID01-dependent mechanism, and that the combination of metabolic intervention and immunotherapy of this pathway might be a promising strategy for the treatment of triple-negative breast cancer (TNBC), and the GCH1 inhibitor could be used as an analgesic45. Members of the GABARAP family (GABARAP, GABARAPL1/GEC1 and GABARAPL2/GATE-16) are one of the subfamilies of the ATG8 protein family, which are related to the receptor and autophagy pathway46. The high-expression of GABARAP is related to the good prognosis of tumors47. CHAC1 is an enzyme related to the activity of γ-glutamyl cyclotransferase that can degrade intracellular GSH and promote ferroptosis of tumor cells48, which has been proved to be related to glioma49 and breast cancer50. CAPG is particularly abundant in macrophage expression51, and CAPG had been proved to be related to tumor cell invasion and tumorigenic52. SLC1A4 is one of the members of solute carrier family 1(SLC1), and SLC1A4 is one of the important roles of amino acid transporter53. SLC1A4 is highly expressed in pancreatic ductal adenocarcinoma and liver cancer cells, and some studies have suggested that SLC1A4 may promote the process of ferroptosis54. ACSL4, a long-chain fatty acyl coenzyme, is closely related to the proliferation and migration of tumor cells55. ACSL4 had been shown to be overexpressed in breast cancer56, GC57, and liver cancer58. ACO1(Cytoplasmic aconitic acid hydratase) is a protein that participates in cytoplasmic and mitochondrial metabolism and, when down regulated, leads to cell death59. NOX is a family of encoded oxidases, NOX4 is a catalytic subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, and NOX5 mainly encodes calcium-dependent NADPH oxidase, produces superoxide, and acts as a calcium-dependent proton channel. The ROS produced by NOX4 is involved in a variety of biological functions, including signal transduction, cell differentiation and tumor cell growth60,61, and NOX4 plays an important role in the process of ferroptosis62. Inhibition of NOX4 can significantly block ferroptosis63. MiRNA plays an important role in tumors. MiR-9 is overexpressed in lung cancer tissues64, and MiR-9 acts as a biomarker for poor prognosis in lung cancer and thyroid papillary carcinoma65. There is no report about the relationship between TUBE1 and tumor in the literature.
Survival analysis indicated that whether in training cohort or the validation cohort, the established risk signature showed effective predictive performance for the survival of GC patients. The ROC curve showed the reliability and stability of the risk signature. And the high risk groups were accompanied with higher Stromal score, higher ESTIMATE score, and lower Tumor Purity. The HPA database examined the IHC staining of FRPGs, and found that the protein expressions of ZFP36, TUBE1, NFE2L2, GCH1, GABARAPL2, CHAC1, CAPG, ACSL4, ACO1, and SLC1A4 in GC and normal tissues were significantly different. Finally, a nomogram integrating the risk score and clinical features was also established and calibrated, and it showed considerable property for predicting the survival. All these results confirmed the prognostic prediction role of FRPGs in GC and correlation between FRPGs and TIME.
Finally, functional analysis was performed to explore potential biological mechanisms. GO enrichment analysis showed that the biological process of DEGs in training cohort mainly included immune globulin, and human immune response mediated by circulating immune globulin. However, the detailed relationship between ferroptosis and immunity is still unclear. Therefore, we performed GSEA analysis to further elucidate the underlying mechanisms, and the results showed that compared with Cluster C2, Cluster C1 showed lower expression in lipid metabolism, which might be related to the poor prognosis of GC patients. These results suggested that the down regulated lipid metabolism resulted in the impairment of TIME, thereby leading to the poor prognosis in GC. Ferroptosis is a regulated oxidative form of cell death associated with the accumulation of lipid ROS due to enhanced lipid peroxidation66. Since Cluster C1 was associated with relatively low levels of ROS, a small proportion of cells die from ferroptosis. Therefore, Cluster C1 had a poor prognosis. Researchers identified that ferroptosis was related to the immune response process67. It is unclear whether and how ferroptosis is involved in T cell immunity and cancer immunotherapy. Studies have shown that immunotherapy-activated CD8 + T cells enhance the specific lipid peroxidation of ferroptosis in tumor cells and that increased ferroptosis contributes to the anti-tumor efficacy of immunotherapy68. CD8 + T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Clinically, tumor ACSL4 correlates with T cell signatures and improved survival in ICB-treated cancer patients69.
KEGG enrichment analysis showed that DEGs in training cohort were mainly enriched in the p53 signaling pathway, IL-17 signaling pathway, MAPK signaling pathway, and PI3K-Akt signaling pathway. Studies have found that these pathways are associated with immune response70,71,72,73. p53 is a tumor suppressor gene, and p53 mutations have been reported in many cancers74. When p53 mutations occur, cells proliferate abnormally and transform into cancer cells. GC patients with p53 mutation have worse prognosis than those without mutation75. More and more evidences support the pathogenic role of IL-17 in cancer formation, including colon cancer76 and lung cancer77. Wu78 found that IL-17 could promote tumor angiogenesis by mediating the up-regulation of VEGF in GC through STAT3 pathway. It has been confirmed that MAPK and PI3K-Akt pathways are involved in many processes of the occurrence and development of GC79,80,81.
Although multiple studies have established relevant prognostic models for ferroptosis in GC82,83, our study shows unique advantages compared with previous studies. Firstly, the number of patients was significantly different from that of published article. Secondly, our work focused on ferroptosis in patients with GC and identified two significantly different molecular subtypes of prognosis and immune status by consensus clustering. Thirdly, genes were obtained in different ways, we have selected differential genes based on molecular subtypes and partially elucidated the underlying mechanisms. Fourthly, we used the GEO data set to validate the prognosis model. Fifthly, we elucidated the effects of ferroptosis on TIME and prognosis. Sixthly, we verified the mRNA expression of FRPGs by qRT-PCR. Seventhly, the prognostic model based on FRPGs we constructed different from the previous articles. Finally, we speculated that the down regulated lipid metabolism may result in the impairment of TIME, thereby leading to the poor prognosis in GC. The establishment of prognostic risk signature based on FRPGs provided new possibilities for us to predict the efficacy of immunotherapy, and promotes personalized treatment for GC patients in the future. However, this study has certain limitations. Our signature was constructed and validated based on retrospective data, without relevant clinical experimental verification.
Conclusion
In summary, in this study, we identified two molecular subtypes, Clusters C1 and C2. In Cluster C1, patients with poor prognosis present with a hyperimmune state and low lipid metabolism, and we speculated that Cluster C1 was in accordance with the immune rejection type. The risk model based on FRPGs can accurately predict the prognosis of GC. These results indicated that ferroptosis is associated with TIME, and the down regulated lipid metabolism may result in the impairment of TIME, thereby leading to the poor prognosis in GC.
Data availability
The data used to support the findings of this study are included within the article.
Abbreviations
- TCGA::
-
The Cancer Genome Atlas
- GEO::
-
Gene Expression Omnibus
- GC::
-
Gastric cancer
- FRGs::
-
Ferroptosis-related genes
- GSEA::
-
Gene set enrichment analysis
- FRPGs::
-
Ferroptosis-related prognosis genes
- TIME::
-
Tumor immune microenvironment
- TIIC::
-
Tumor infiltrating immune cells
- HPA::
-
Human protein atlas
- IHC::
-
Immunohistochemical
- DEGs::
-
Differentially expressed genes
- GO::
-
Gene ontology
- BP::
-
Biological process
- CC::
-
Cellular component
- MF::
-
Molecular function
- FDR::
-
Error detection rate
- KEGG::
-
Kyoto Encyclopedia of Genes and Genomes
- PPI::
-
Protein–protein interaction
- OS::
-
Overall survival
References
Song, S. Y. SP: Development and verification of ferroptosis-related gene signature for predicting the prognosis and immune microenvironment in gastric cancer (2021).
Ferlay, J. et al. Cancer statistics for the year 2020: An overview. Int. J. Cancer 149, 778–789 (2021).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249 (2021).
Zheng, S. et al. Identification of the angiogenesis related genes for predicting prognosis of patients with gastric cancer. BMC Gastroenterol. 21(1), 146 (2021).
Dixon, S. J. et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149(5), 1060–1072 (2012).
Zhao, Y. et al. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 16(1), 79 (2017).
Lai, Y. et al. STYK1/NOK correlates with ferroptosis in non-small cell lung carcinoma. Biochem. Biophys. Res. Commun. 519(4), 659–666 (2019).
Ding, Y. et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J. Hematol. Oncol. 14(1), 19 (2021).
Chen, P. et al. Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 10(11), 5107–5119 (2020).
Zhao, L. et al. Apatinib induced ferroptosis by lipid peroxidation in gastric cancer. Gastric Cancer 24(3), 642–654 (2021).
Chen, X. et al. Identification of a ferroptosis-related signature associated with prognosis and immune infiltration in adrenocortical carcinoma. Int. J. Endocrinol. 2021, 4654302 (2021).
Qiu, C. J. et al. Development and validation of a ferroptosis-related prognostic model in pancreatic cancer. Invest. New Drugs 39(6), 1507–1522 (2021).
Hsieh, C. H. et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics 11(14), 7072–7091 (2021).
Xu, F. et al. The roles of ferroptosis regulatory gene SLC7A11 in renal cell carcinoma: A multi-omics study. Cancer Med. 10(24), 9078–9096 (2021).
Babjuk, M. et al. European association of urology guidelines on non-muscle-invasive bladder cancer (Ta, T1, and carcinoma in situ). Eur. Urol. 81, 75–94 (2021).
Sylvester, R. J. et al. European Association of Urology (EAU) Prognostic Factor Risk Groups for Non-muscle-invasive Bladder Cancer (NMIBC) Incorporating the WHO 2004/2016 and WHO 1973 classification systems for grade: An update from the EAU NMIBC guidelines panel. Eur. Urol. 79(4), 480–488 (2021).
Joshi, S. S. & Badgwell, B. D. Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 71(3), 264–279 (2021).
Dashti, A., Ebrahimi, M., Hadjati, J., Memarnejadian, A. & Moazzeni, S. M. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 374(1), 175–185 (2016).
Kono, K., Nakajima, S. & Mimura, K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer 23(4), 565–578 (2020).
Schneider, A. K., Chevalier, M. F. & Derre, L. The multifaceted immune regulation of bladder cancer. Nat. Rev. Urol. 16(10), 613–630 (2019).
Xue, Y. et al. Tumorinfiltrating M2 macrophages driven by specific genomic alterations are associated with prognosis in bladder cancer. Oncol. Rep. 42(2), 581–594 (2019).
Lee, D. A. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol. Rev. 290(1), 85–99 (2019).
Fernandes, J. V. et al. The role of the mediators of inflammation in cancer development. Pathol. Oncol. Res. 21(3), 527–534 (2015).
Johnston, M. P. & Khakoo, S. I. Immunotherapy for hepatocellular carcinoma: Current and future. World J. Gastroenterol. 25(24), 2977–2989 (2019).
Zhou, N. & Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database (Oxford) https://doi.org/10.1093/database/baaa021 (2020).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77(21), e108–e110 (2017).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015).
Engebretsen, S. & Bohlin, J. Statistical predictions with glmnet. Clin. Epigenet. 11(1), 123 (2019).
Shi, X. et al. Next-generation sequencing identifies novel genes with rare variants in total anomalous pulmonary venous connection. EBioMedicine 38, 217–227 (2018).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10(1), 1523 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49(D1), D545–D551 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Lv, Z. et al. Identification of a prognostic signature based on immune-related genes in bladder cancer. Genomics 113(3), 1203–1218 (2021).
Hinshaw, D. C. & Shevde, L. A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79(18), 4557–4566 (2019).
Gambardella, V. et al. The role of tumor-associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat. Rev. 86, 102015 (2020).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541(7637), 321–330 (2017).
Salmon, H. et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 122(3), 899–910 (2012).
Qin, X. et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 17, 4266–4285 (2021).
Zhang, Z. et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 16(8), 1482–1505 (2020).
Hu, X. F. et al. Nrf2 overexpression predicts prognosis and 5-FU resistance in gastric cancer. Asian Pac. J. Cancer Prev. 14(9), 5231–5235 (2013).
Kawasaki, Y. et al. Clinicopathological significance of nuclear factor (erythroid-2)-related factor 2 (Nrf2) expression in gastric cancer. BMC Cancer 15, 5 (2015).
Wei, J. L. et al. GCH1 induces immunosuppression through metabolic reprogramming and IDO1 upregulation in triple-negative breast cancer. J. Immunother. Cancer 9(7), e002383 (2021).
Pickert, G. et al. Inhibition of GTP cyclohydrolase reduces cancer pain in mice and enhances analgesic effects of morphine. J. Mol. Med. (Berl.) 90(12), 1473–1486 (2012).
Hervouet, E. et al. The autophagy GABARAPL1 gene is epigenetically regulated in breast cancer models. BMC Cancer 15, 729 (2015).
Jacquet, M., Guittaut, M., Fraichard, A. & Despouy, G. The functions of Atg8-family proteins in autophagy and cancer: Linked or unrelated?. Autophagy 17(3), 599–611 (2021).
Chen, M. S. et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 8(70), 114588–114602 (2017).
Chen, P. H. et al. The CHAC1-inhibited Notch3 pathway is involved in temozolomide-induced glioma cytotoxicity. Neuropharmacology 116, 300–314 (2017).
Jahn, B. et al. Development of a novel prognostic score for breast cancer patients using mRNA expression of CHAC1. J. Comp. Eff. Res. 6(7), 563–574 (2017).
Yu, F. X., Johnston, P. A., Sudhof, T. C. & Yin, H. L. gCap39, a calcium ion- and polyphosphoinositide-regulated actin capping protein. Science 250(4986), 1413–1415 (1990).
Pellieux, C. et al. Cap G, a gelsolin family protein modulating protective effects of unidirectional shear stress. J. Biol. Chem. 278(31), 29136–29144 (2003).
Kanai, Y. & Hediger, M. A. The glutamate/neutral amino acid transporter family SLC1: Molecular, physiological and pharmacological aspects. Pflugers Arch. 447(5), 469–479 (2004).
Dixon, S. J. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014).
Cao, Y., Dave, K. B., Doan, T. P. & Prescott, S. M. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 61(23), 8429–8434 (2001).
Monaco, M. E. et al. Expression of long-chain fatty acyl-CoA synthetase 4 in breast and prostate cancers is associated with sex steroid hormone receptor negativity. Transl. Oncol. 3(2), 91–98 (2010).
Ye, X., Zhang, Y., Wang, X., Li, Y. & Gao, Y. Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in gastric cancer. IUBMB Life 68(4), 320–327 (2016).
Sung, Y. K. et al. Fatty acid-CoA ligase 4 is overexpressed in human hepatocellular carcinoma. Cancer Sci. 94(5), 421–424 (2003).
Gonzalez-Sanchez, L. et al. Exploiting the passenger ACO1-deficiency arising from 9p21 deletions to kill T-cell lymphoblastic neoplasia cells. Carcinogenesis 41(8), 1113–1122 (2020).
Guo, S. & Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 23(10), 888–896 (2015).
Lu, J. P. et al. Androgens induce oxidative stress and radiation resistance in prostate cancer cells though NADPH oxidase. Prostate Cancer Prostatic Dis. 13(1), 39–46 (2010).
Yang, W. H. et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 28(10), 2501–2508 (2019).
Poursaitidis, I. et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 18(11), 2547–2556 (2017).
Chen, X. et al. Oncogenic miR-9 is a target of erlotinib in NSCLCs. Sci. Rep. 5, 17031 (2015).
Sondermann, A. et al. MiR-9 and miR-21 as prognostic biomarkers for recurrence in papillary thyroid cancer. Clin. Exp. Metastasis 32(6), 521–530 (2015).
Stockwell, B. R. et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171(2), 273–285 (2017).
Matsushita, M. et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212(4), 555–568 (2015).
Wang, W. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569(7755), 270–274 (2019).
Liao, P. et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40, 365–378 (2022).
Zhang, X. et al. PI3K/AKT/p53 pathway inhibits infectious spleen and kidney necrosis virus infection by regulating autophagy and immune responses. Fish Shellfish Immunol. 120, 648–657 (2022).
Ngan, H. L. et al. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Sci. Alliance 3(6), e201900545 (2020).
Fan, Z. P. et al. S100A9 activates the immunosuppressive switch through the PI3K/Akt pathway to maintain the immune suppression function of testicular macrophages. Front. Immunol. 12, 743354 (2021).
Bianchi, E., Vecellio, M. & Rogge, L. Editorial: Role of the IL-23/IL-17 pathway in chronic immune-mediated inflammatory diseases: Mechanisms and targeted therapies. Front. Immunol. 12, 770275 (2021).
Saxena, A., Shukla, S. K., Prasad, K. N. & Ghoshal, U. C. Analysis of p53, K-ras gene mutation & Helicobacter pylori infection in patients with gastric cancer & peptic ulcer disease at a tertiary care hospital in north India. Indian J. Med. Res. 136(4), 664–670 (2012).
Solcia, E. et al. A combined histologic and molecular approach identifies three groups of gastric cancer with different prognosis. Virchows Arch. 455(3), 197–211 (2009).
Zepp, J. A. et al. IL-17A-induced PLET1 expression contributes to tissue repair and colon tumorigenesis. J. Immunol. 199(11), 3849–3857 (2017).
Jin, C. et al. Commensal microbiota promote lung cancer development via gammadelta T cells. Cell 176(5), 998–1013 (2019).
Wu, X. et al. IL-17 promotes tumor angiogenesis through Stat3 pathway mediated upregulation of VEGF in gastric cancer. Tumour Biol. 37(4), 5493–5501 (2016).
Du, F. et al. DDIT4 promotes gastric cancer proliferation and tumorigenesis through the p53 and MAPK pathways. Cancer Commun. (Lond.) 38(1), 45 (2018).
Wu, S. et al. ORAI2 promotes gastric cancer tumorigenicity and metastasis through PI3K/Akt signaling and MAPK-dependent focal adhesion disassembly. Cancer Res. 81(4), 986–1000 (2021).
Zhang, Q. et al. Berberine represses human gastric cancer cell growth in vitro and in vivo by inducing cytostatic autophagy via inhibition of MAPK/mTOR/p70S6K and Akt signaling pathways. Biomed. Pharmacother. 128, 110245 (2020).
Jiang, X. et al. Construction and validation of a ferroptosis-related prognostic model for gastric cancer. J. Oncol. 2021, 6635526 (2021).
Xiao, S., Liu, X., Yuan, L., Chen, X. & Wang, F. Expression of ferroptosis-related genes shapes tumor microenvironment and pharmacological profile in gastric cancer. Front. Cell Dev. Biol. 9, 694003 (2021).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Nos. 81673918). Pilot GC project of clinical collaboration of traditional Chinese medicine and Western medicine on major difficult diseases in the state administration of traditional Chinese medicine; 2019 “Construction Project of Evidence-based Capacity for Traditional Chinese Medicine” (2019XZZX-ZL003) in state administration of traditional Chinese medicine; Open Program of the Third Phase of the Program of Traditional Chinese Medicine (TCM) Advantageous Subjects (ZYX03KF020); Science and Technology Project of Jiangsu Provincial Administration of Traditional Chinese Medicine (ZD201803).
Author information
Authors and Affiliations
Contributions
P.S. designed the research. S.S. analyzed the data and wrote the paper. All authors read and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, S., Shu, P. Expression of ferroptosis-related gene correlates with immune microenvironment and predicts prognosis in gastric cancer. Sci Rep 12, 8785 (2022). https://doi.org/10.1038/s41598-022-12800-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12800-6
This article is cited by
-
Differential roles of highly expressed PFKFB4 in colon adenocarcinoma patients
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
