
Abstract
The generation of reference data for deep learning models is challenging for reactive systems, and more so for combustion reactions due to the extreme conditions that create radical species and alternative spin states during the combustion process. Here, we extend intrinsic reaction coordinate (IRC) calculations with ab initio MD simulations and normal mode displacement calculations to more extensively cover the potential energy surface for 19 reaction channels for hydrogen combustion. A total of ∼290,000 potential energies and ∼1,270,000 nuclear force vectors are evaluated with a high quality range-separated hybrid density functional, ωB97X-V, to construct the reference data set, including transition state ensembles, for the deep learning models to study hydrogen combustion reaction.
Measurement(s) | ab initio energies and forces of hydrogen combustion |
Technology Type(s) | density functional theory • ab initio molecular dynamics • normal modes |
Factor Type(s) | cartesian coordinates |
Similar content being viewed by others
Background & Summary
The expectation behind training deep learning models to predict molecular energies and atomic forces of molecules is the requirement of large data sets. However, very recently it has become recognized that deep learning methods that are designed with rotationally equivariant operators offer a significant reduction in data needed for training relative to invariant ML models1,2,3,4, and often outcompete even kernal methods that have traditionally been considered advantageous due to their low data requirements5. However, the promise in regards equivariant deep learning models must be further validated by construction of more challenging data sets than encountered up until now. For example, the recent SN2 data set provides reference energy and forces for more than 450,000 structures calculated using Density Functional Theory (DFT), but ultimately is data on highly similar individual reactions of methyl halides with one of four substituted halogens, F, Cl, Br, and I6.
Capturing the energy release in hydrogen combustion is a proposed energy solution for zero CO2 emissions, and many of the elementary reactions of H2 combustion are also present in other types of fuel generation7. Under realistic reaction conditions of very high temperature and high pressure make it extremely difficult to study H2 combustion reactions experimentally. Because hydrogen combustion is difficult to study experimentally under these extremes8, theoretical models must play an active role in filling the breach, but fundamentally relies on an accurate potential energy model of not only the elementary reactions9 but the excursions away from the reaction coordinate.
Hydrogen combustion, despite being the simplest combustion system, is nonetheless still quite chemically complicated because it can encounter one or more 19 reaction channels during the combustion event depending on the physical conditions of high temperatures and pressures8. This compounds the need for high quality data that is expensive to generate given the need for extensive sampling and the presence of metastable points such as transition states. For non-reacting chemical systems, conventional MD simulations are well-suited for generating a large number of configurations, which are then used as input into single point quantum-chemical energy and force calculations10,11,12. However, for reactive systems, conventional force-field based MD simulations are not useful as they don’t allow breaking and forming of chemical bonds. Recent work has attempted to address this deficiency through graph-based methods that generate reference data for reactive systems13,14, but they are also prone to produce large numbers of specious chemical states and unrealistic intermediates such as highly unstable radicals. Therefore fully ab initio sampling methods are a necessity for creation of the many molecular fragments involved in combustion chemistry, including the presence of stable and unstable intermediates, high energy transition states, and a variety of product molecules that can be formed during the reaction that is dependent on the reactive channel8,9,15,16,17,18.
Our goal here is to characterize the potential energy surface (PES) of hydrogen combustion through the reaction channels proposed by Li et al.19 using a systematic approach in ab initio data generation that samples off the intrinsic reaction coordinate (IRC). This study provides a data set of ∼290,000 potential energies and ∼1,270,000 nuclear force vectors for structures that are sampled near and far from the IRC for 19 hydrogen combustion sub-reactions, some of which are barrierless transitions, others are dominated by large activation barriers, and even reactions involving changes in spin state19. This data set offers a new ML benchmark set that allows systematic investigation of data reduction when using emerging equivariant deep learning model, as well as being of interest in its own right as a source of data for machine learning of energy and forces that drive an MD engine for combustion under extreme thermodynamic conditions.
Methods
We have used fully ab initio methods for sampling 19 reactive channels for hydrogen combustion as summarized in Table 1. For each reaction we used the ωB97X-V DFT functional20 with the cc-pVTZ basis set. All calculations were performed as unrestricted open shell, using an ultrafine integration grid of 99 radial points and 590 angular points, with an SCF convergence of \(1{0}^{-8}\) using the GDM method21. All potential energies for each configuration of the 19 reactions are reported as ΔE
using the atomic energies EH = −0.5004966690 a.u. and Eo = −75.0637742413 a.u., and with ΔE converted to units of kcal/mole. All calculations were performed using the Q-Chem program22,23.
We have organized the PES data into four categories that classify the reaction mechanism involved in the elementary steps for each reactive channel: association/dissociation reactions (channels 5-9 and 15), substitution reactions (channel 16), oxygen transfer (channels 1, 11, and 12), and hydrogen transfer (channels 2-4, 10, 13, 14, 17–19). We have kept the same numbering scheme as Li and co-workers19 in these categories so that readers can refer back to any particular IRC of that work if desired.
The PES for each reaction channel are visualized by means of two collective variables of coordination numbers (CN) represented by
where \({r}_{0}\) is the equilibrium distance and \(\sigma =3.0\) controls the sharpness of the function. Reaction channels 5–7 involve only two atoms, and thus only a 1-D distance scan is performed.
Finally, we developed a strategy for extensive sampling of the PES for the 19 reaction channels for hydrogen combustion as follows:
-
1.
Transition States and IRCs. Approximate TS structures were found using the freezing string method24,25, and refined by the partitioned-rational function optimization eigenvector following method (P-RFO)26. An IRC scan is then generated, and vibrational frequency analysis was performed to confirm that reactants and products have no imaginary frequencies and the TS has only one imaginary frequency. As the IRC configurations connect the minimum energy pathway, and therefore span a meaningful fraction of the configurational space of a given reaction, they serve as useful starting geometries for systematic normal mode displacements and stochastic generation of structures using AIMD at finite temperatures to explore the PES for each reaction channel in more detail.
-
2.
AIMD Simulations. We employed AIMD simulations to sample configurations around the IRC structures using the TS as the initial configuration for each of the reaction channels. The AIMD simulations were performed at four different high temperatures by initializing the Maxwell-Boltzmann distribution of velocities at temperatures of 500 K, 1000 K, 2000 K and 3000 K. Furthermore at each temperature three different simulation timescales are performed using a 1.21 fs (1.au.) time step: 10 independent (i.e. reinitialized velocities) long simulations of 121 fs, 20 independent short trajectories of 60.5 fs, and finally 25 very short simulations of 24.2 fs. In summary, the AIMD calculations yielded a total of 10000 configurations along with their potential energies and nuclear forces for each reaction channel (see Table 1).
-
3.
Normal Mode Displacements. Systematic normal mode displacements along the IRC is performed. Starting from each IRC structure, the frequencies were calculated and all atoms were displaced along each normal mode (NM) with a \(\pm 0.01\), \(\pm 0.025\), \(\pm 0.05\), \(\pm 0.075\), \(\pm 0.1\), \(\pm 0.125\), and ±0.15. increment. These sampled structures that compress or expand the IRC structures help to diversify the AIMD geometries for each reaction, yielding ∼ 127,000 configurations as summarized in Table 1. The IOData Python library was used for parsing the Q-Chem output files in generating these geometries27.
Technical Validation
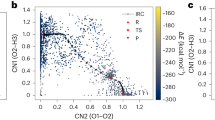
Figure 1 provides a representative ab initio sampling of one of the hydrogen transfer reactions, \({\rm{O}}+{H}_{2}\to OH+H\), in which two collective coordinates reasonably capture the potential energy surface of this reaction channel. Upon analyzing the AIMD generated geometries and their energies, it is noticed that both the reactant and product endpoint regions are well sampled (Fig. 1(a)). However, near the transition state or in regions of high slope on the potential energy surface, data points from the AIMD simulations are more sparse. The addition of normal mode displacement points greatly improves sampling the configuration space of the PES along the IRC path (Fig. 1(b)).

Potential energy surface for the hydrogen transfer reaction 2 (\({\rm{O}}+{{\rm{H}}}_{2}\to {\rm{OH}}+{\rm{H}}\)). (a) showing IRC and AIMD sample data only and (b) including normal mode data. CN1 represents the breaking of the H-H bond and CN2 represents the formation of the O-H bond. All energies are reported with respect to the atomization energies as given in Eq. (1) in units of kcal/mol. The red dots on the energy surface are configurations with energies larger than 10 kcal/mol of the energy of the TS structure. The points denoted with R, TS and P are corresponds to the reactant, transition state and product, respectively.
Figure 2 shows that the AIMD and NM calculations are complementary for sampling different areas away from the IRC, particularly evident for reaction channel 1 involving oxygen transfer (Fig. 2(a)), reaction 8 that probes the association reaction mechanism (Fig. 2(b)), and for reaction channel 16 pertaining to a substitution mechanism (Fig. 2(c)). In all cases the use of two collective coordinates is sufficient to capture the IRC and its AIMD and NM extensions, borne out in the supplementary information Figures S1–S4 that provides the potential energy surfaces generated for the remaining reaction channels for these classes of hydrogen combustion reactions.

Representative potential energy surfaces for oxygen transfer, association, and substitution reactions along two reaction coordinates CN1 and CN2. (a) oxygen transfer reaction 1 (\({\rm{H}}+{{\rm{O}}}_{2}\to {\rm{O}}{\rm{H}}+{\rm{O}}\)), (b) association reaction 8 (\({\rm{H}}+{\rm{O}}{\rm{H}}\to {{\rm{H}}}_{2}{\rm{O}}\)), and (c) substitution reaction 16 (\({{\rm{H}}}_{2}{{\rm{O}}}_{2}+{\rm{H}}\to {{\rm{H}}}_{2}{\rm{O}}+{\rm{OH}}\)). Each CN represents the formation or breaking of respective bond involved in the reaction process mentioned in the axes.
Figure 3 shows the nature of the alternative potential energy surfaces that are represented by the changes in spin state from doublet to quartet for the oxygen transfer reaction channel 12. Figure 3(a) shows that the energy difference between the two spin states is very small near the reactant, less than 0.2 kcal/mol, but favors the quartet state substantially around the product. Figure 3(b) plots the IRC using either the doublet or quartet spin state energies using the quartet spin state static structures, and similarly Fig. 3(c) represents the two spin state energies using the doublet energy configurations. Figure 3(d) shows the minimum energy of the two spin states along a single generated IRC. These differences indicate that while the geometric effects may be small, the electronic energy differences between spin states are significant. In the supplementary information we also provides the potential energy surfaces generated for reaction channel 6 which also undergoes a spin state change.

The changes in the PES for reaction channel 12 involving changes in spin state. (a) the spin cross over between the two closely spaced doublet and quartet spin state energy levels around the reactant region with widening differences progressing to product. (b) the IRC path defined by the doublet energy but geometries from the quartet (green), and from the doublet energy and geometries (red). (c) the IRC path defined by the quartet energy but geometries from the doublet (green) and from the quartet energies and geometries (red). (d) Resultant PES obtained reaction channel 12 (\({{\rm{HO}}}_{2}+{\rm{O}}\to {\rm{OH}}+{{\rm{O}}}_{2}\)) by choosing the minimum energy between the two spin states. Each CN represents the formation or breaking of respective bond involved in the reaction process mentioned in the axes.
In summary, we generated high quality DFT data for hydrogen combustion reaction channels using range separated hybrid meta-GGA functional ωB97X-V with the cc-pVTZ basis set. This level of theory is considered highly accurate for thermochemistry and reactive barriers28,29, and the IRC profiles compared against the gold standard CCSD(T)/cc-pVTZ methods determined very small errors with the DFT level of theory7. This work moves beyond benchmarks such as the IRC for H2 combustion by extensive sampling off the reaction coordinate using ab initio MD simulation and normal mode analysis for each of the 19 reaction channels. Furthermore, we also consider multiple spin states of the species formed in the hydrogen combustion process. This high quality data is now available to benchmark deep learning models for chemical reactivity, and as a model of the PES for generating kinetic models of H2 combustion, especially at high pressure.
Data Records
All data can be found in the figshare repository. For each reaction channel the IRC, AIMD and NM generated configurations and corresponding energies and atomic forces are provided in.npz file format; for reaction channel 5, 6 and 7 only IRC generated data are provided as discussed above. Each .npz file contains six keys including, “R” (atomic Cartesian coordinates), “Z” (atomic numbers), “N” (number of atoms), “ΔE” (reference potential energy), “F” (atomic force vectors), and “RXN” (reaction number). All the atomic position are in Å and energy and force vectors are provided in kcal/mol and kcal/mol/Å, respectively. Reaction channels such as 6 and 12 involve nuclear spin changes during the reaction, and therefore IRC calculations are performed for both spin states, with the data sorted to either (1) retain energies and forces consistent with one spin state, or (2) retaining the lowest energy spin state along the IRC for each channel. Furthermore, for reactions 6 and 12 two sets of data are provided namely 06a/06b and 12a/12b corresponding to two different spin states involved in the reaction process.
Usage Notes
The data set contains 19 folders corresponding to each of the reaction channels. Each reaction channel has three.npz files storing the geometries and corresponding potential energies energies and atomic force vectors obtained from IRC, AIMD and NM simulations separately. Each.npz file contains the “R” (atomic Cartesian coordinates), “Z” (atomic numbers), “N” (number of atoms), “ΔE” (reference potential energy), “F” (atomic forces), and “RXN” (reaction number) keys and the corresponding values for each configuration.
Code availability
All the data and python scripts used to generate coordination number based PES surface to analyze the data for each reaction channel is provided at https://doi.org/10.6084/m9.figshare.1960168930.
References
Batzner, S. et al. Se(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. arXiv preprint arXiv:2101.03164, 2021.
Schütt, K. T. et al. Equivariant message passing for the prediction of tensorial properties and molecular spectra. arXiv preprint arXiv:2102.03150, 2021.
Qiao, Z. et al. Unite: Unitary n-body tensor equivariant network with applications to quantum chemistry. arXiv preprint arXiv:2105.14655, 2021.
Haghighatlari, M. et al. Newtonnet: A newtonian message passing network for deep learning of interatomic potentials and forces. arXiv preprint arXiv:2108.02913, 2021.
Haghighatlari, M., et al. Learning to make chemical predictions: The interplay of feature representation, data, and machine learning methods. Chem, 6 (7): 1527–1542, ISSN 2451-9294. https://doi.org/10.1016/j.chempr.2020.05.014 2020.
Unke, O. T. & Meuwly, M. PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. J. Chem. Theory Comput. 15(6), 3678–3693, https://doi.org/10.1021/acs.jctc.9b00181 (2019).
L. W. Bertels, L. B. Newcomb, M. Alaghemandi, J. R. Green, and M. Head-Gordon. Benchmarking the Performance of the ReaxFF Reactive Force Field on Hydrogen Combustion Systems. J. Phys. Chem. A, 124(27), 5631–5645, ISSN 15205215, https://doi.org/10.1021/acs.jpca.0c02734 (2020).
Li, J., Zhao, Z., Kazakov, A. & Dryer, F. An updated comprehensive kinetic model of hydrogen combustion. International Journal of Chemical Kinetics 36, 566–575, https://doi.org/10.1002/kin.20026 (2004).
Grambow, C., Pattanaik, L. & Green, W. Reactants, products, and transition states of elementary chemical reactions based on quantum chemistry. Scientific Data 7, 137, https://doi.org/10.1038/s41597-020-0460-4 (2020).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401, https://doi.org/10.1103/PhysRevLett.98.146401 (2007).
Smith, J. S., Isayev, O. & Roitberg, A. E. Ani-1: an extensible neural network potential with dft accuracy at force field computational cost. Chemical Science 8(4), 3192–3203 (2017).
St. John, P. et al. Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules. Scientific Data 7, 244, https://doi.org/10.1038/s41597-020-00588-x (2020).
Margraf, J. & Reuter, K. Systematic enumeration of elementary reaction steps in surface catalysis. ACS Omega 4, 3370–3379, https://doi.org/10.1021/acsomega.8b03200 (2019).
Stocker, S., Csányi, G., Reuter, K. & Margraf, J. Machine learning in chemical reaction space. Nature Communications 11, 10, https://doi.org/10.1038/s41467-020-19267-x (2020).
Gerasimov, G. & Shatalov, O. Kinetic mechanism of combustion of hydrogen–oxygen mixtures. Journal of Engineering Physics and Thermophysics 86, 987–995, https://doi.org/10.1007/s10891-013-0919-7 (2013).
Simm, G. & Reiher, M. Context-driven exploration of complex chemical reaction networks. Journal of Chemical Theory and Computation 13, 09, https://doi.org/10.1021/acs.jctc.7b00945 (2017).
Ulissi, Z., Medford, A., Bligaard, T. & Nørskov, J. To address surface reaction network complexity using scaling relations machine learning and dft calculations. Nature Communications 8, 14621, https://doi.org/10.1038/ncomms14621 (2017).
Zeng, J., Cao, L., Xu, M., Zhu, T. & Zhang, J. Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation. Nature Communications 11, 5713, https://doi.org/10.1038/s41467-020-19497-z (2020).
J. Li, Z. Zhao, A. Kazakov, and F. L. Dryer. An updated comprehensive kinetic model of hydrogen combustion. International Journal of Chemical Kinetics, 36(10), 566–575, https://doi.org/10.1002/kin.20026 2004.
Mardirossian, N. & Head-Gordon, M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 16, 9904–9924, https://doi.org/10.1039/c3cp54374a (2014).
Van Voorhis, T. & Head-Gordon, M. A geometric approach to direct minimization. Molecular Physics 100(11), 1713–1721, https://doi.org/10.1080/00268970110103642 (2002).
Shao, Y. et al. Advances in molecular quantum chemistry contained in the q-chem 4 program package. Molecular Physics 113(2), 184–215, https://doi.org/10.1080/00268976.2014.952696 (2015).
Epifanovsky, E. et al. Software for the frontiers of quantum chemistry: An overview of developments in the q-chem 5 package. The Journal of Chemical Physics 155(8), 084801 (2021).
Behn, A., Zimmerman, P., Bell, A. & Head-Gordon, M. Efficient exploration of reaction paths via a freezing string method. The Journal of chemical physics 135, 224108, https://doi.org/10.1063/1.3664901 (2011).
Mallikarjun Sharada, S., Zimmerman, P., Bell, A. & Head-Gordon, M. Automated transition state searches without evaluating the hessian. Journal of Chemical Theory and Computation 8, 5166–5174, https://doi.org/10.1021/ct300659d (2012).
Baker, J. An algorithm for the location of transition states. Journal of Computational Chemistry 7, 385–395 (1986).
T. Verstraelen et al. Iodata: A python library for reading, writing, and converting computational chemistry file formats and generating input files. Journal of Computational Chemistry, 42 (6): 458–464, https://doi.org/10.1002/jcc.26468. onlinelibrary.wiley.com/doi/abs/10.1002/jcc.26468 2021.
Mardirossian, N. & Head-Gordon, M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Molecular Physics 115(19), 2315–2372, https://doi.org/10.1080/00268976.2017.1333644 (2017).
Goerigk, L. et al. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 19, 32184–32215, https://doi.org/10.1039/C7CP04913G (2017).
Guan, X. et al. Hydrogen Combustion using IRC, AIMD and normal modes. Figshare https://doi.org/10.6084/m9.figshare.19601689 (2022).
Acknowledgements
We thank the National Science Foundation under grant CHE-1955643. F.H-Z. acknowledges financial support from Natural Sciences and Engineering Research Council (NSERC) of Canada. M. Liu thanks the China Scholarship Council for a visiting scholar fellowship. C.J.S. acknowledges funding by the Ministry of Innovation, Science and Research of North Rhine-Westphalia (“NRW Rückkehrerprogramm”) and an Early Postdoc Mobility fellowship from the Swiss National Science Foundation. This research used computational resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
X.G., A.D., C.J.S., F.H.-Z., L.B., M.H., M.H.-G. and T.H.-G. conceived the scientific direction for the hydrogen combustion data set, and wrote the complete manuscript. All authors provided comments on the results and manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guan, X., Das, A., Stein, C.J. et al. A benchmark dataset for Hydrogen Combustion. Sci Data 9, 215 (2022). https://doi.org/10.1038/s41597-022-01330-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01330-5
This article is cited by
-
Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential
Nature Chemistry (2024)
-
Using machine learning to go beyond potential energy surface benchmarking for chemical reactivity
Nature Computational Science (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}