
Abstract
Prenatal exome sequencing could be complex because of limited phenotypical data compared to postnatal/portmortem phenotype in fetuses affected by multiple congenital abnormalities (MCA). Here, we investigated limits of prenatal phenotype for ES interpretation thanks to a blindly reanalysis of postmortem ES data using prenatal data only in fetuses affected by MCA and harboring a (likely)pathogenic variant or a variant of unknown significance (VUS). Prenatal ES identified all causative variant previously reported by postmortem ES (22/24 (92%) and 2/24 (8%) using solo-ES and trio-ES respectively). Prenatal ES identified 5 VUS (in four fetuses). Two of them have been previously reported by postmortem ES. Prenatal ES were negative for four fetuses for which a VUS were diagnosed after autopsy. Our study suggests that prenatal phenotype is not a limitation for implementing pES in the prenatal assessment of unsolved MCA to personalize fetal medicine and could influence indication of postmortem examination.
Similar content being viewed by others

Introduction
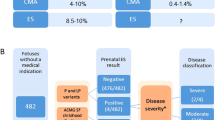
Congenital abnormalities (CA) occur in 2–5% of pregnancies and are the main cause of perinatal death (20–25%) [1]. Detecting CA, especially to identify genetic disorders that affect fetal prognosis, is one of the biggest challenges in prenatal care [2]. Current prenatal genetic assessment of fetal malformations based on standard karyotype and chromosomal micro-array analysis (CMA) identifies chromosomal abnormalities and pathogenic copy number variants (CNV) in approximately 20% and 6% of cases, respectively [3]. Identification of the causal genetic abnormalities is required for diagnosis; to adapt prenatal/perinatal management according to the prognosis; and to provide genetic counseling for the current pregnancy, and any subsequent pregnancies [4]. In negative cases (about 70%), the management is limited due to remaining challenges relative to genetic counseling [5].
In the last decade, several studies have highlighted the contribution of next-generation sequencing, particularly exome sequencing (ES), in the investigation of postnatally suspected monogenic diseases, with a mean diagnostic yield of approximately 40% [6]. The use of ES to investigate CA was progressively extended from postmortem to prenatal diagnosis. It has been illustrated that the diagnostic yield seems to be lower than for other postnatal indications (i.e intellectual disability), with an overall diagnostic yield of approximately 20%. However, the diagnostic value of pES in CA should no longer be controversial, with a diagnostic yield greater than the additive value of CMA relative to karyotype [3]. However, a high variation has been reported (from 2 to 71%) depending on the series’ inclusion criteria and the methodology used [4, 7, 8].
Although the diagnostic yield of prenatal ES (pES) appears to be lower, running prenatal genetic tests allows parents to obtain additional information about the disease and prognosis, and may influence pregnancy outcome and future family planning [9]. Implementation of pES in clinical practice could be limited by cost, turnaround time, interpretation based on a “partial” fetal phenotype [4], incidental findings, and management of variants of unknown significance (VUS).
In case of lethal CA, postmortem phenotype, mostly report in clinical databases, with potential additional information provided by prenatal imaging, is considered as the reference for genetic investigations. It could be reconsidered by improvement of prenatal imaging, parental reluctance for postmortem examination and development of genomic medicine.
To investigate limits of prenatal phenotype for ES interpretation, we compared results of ES analyzed using prenatal data only versus postmortem clinical data.
Material and methods
Affected cases
We performed an ancillary study from the collaborative French cohort including 95 fetuses with lethal and unsolved multisystemic CA investigated by postmortem solo-ES (NCT02512354-FOETEX) [10]. Briefly, fetuses included after termination of pregnancy (TOP), intrauterine or neonatal death, had at least two congenital malformations and no etiological clinical diagnosis after fetal examinations and current investigations (CMA, targeted sequencing). For the present study, we included all fetuses with a likely pathogenic or a pathogenic (LP/P) variant or a VUS identified by postmortem solo-ES. In the FOETEX study, clinical data available for the ES analysis were almost exclusively from the postmortem examination, the family history and the risk factors (consanguinity, previous TOP, intrauterine fetal death…); prenatal data were not detailed. Parents provided written informed consent and the study was approved by the local ethics committee.
Prenatal clinical data and ES analysis
We retrospectively collected all prenatal data in a standardized phenotyping report, especially fetal imaging reports (ultrasound, CT-scan, MRI), biological investigations (Down syndrome screening, tests performed on chorionic villus sampling or amniotic fluid), risk factors (toxic or work-related exposure…) and obstetrical diseases. Prenatal data were compiled in a standardized report by a specialist in fetal imaging without knowledge of the pregnancy outcome.
ES data were blindly reanalyzed by biologists that did not have previously participated in the initial ES analysis based on fetal autopsy. pES analysis was performed using a sequential approach: first, a solo-pES analysis to limit costs, using the multistep methodology previously described by Lefebvre et al., and second a trio-ES (trio-pES) for negative cases. To compare pES and postmortem ES, the analysis of pES data was made thanks to the pipeline previously used for the original study (Lefebvre et al.,) without updating of annotation and databases. Candidate variants were multidisciplinary discussed and classified following the American College of Medical Genetics and Genomics recommendations [11]. Secondary and incidental findings were not allowed.
Results
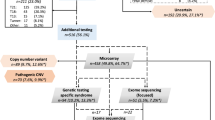
A total of 32 fetuses (17 males, 15 females) were included, involving 24 cases with a causal diagnosis and eight cases harboring a VUS after postmortem solo-ES. Clinical data are summarized in Table 1.
Solo-pES identified a LP/P variant in 22 fetuses, all of them previously identified by postmortem ES (Table 1).
Subsequent trio-pES identified the causal diagnoses for the two last cases solved by postmortem ES (F.10 and F.17), increasing the diagnostic yield from 92% (22/24) to 100%, and 5 VUS in 4 fetuses. For F.25 with a phenotype suggestive of a congenital bone disorder, a VUS in INPPL1 and COL1A1 was respectively identified by prenatal and postmortem ES. For two cases with a syndromic congenital diaphragmatic hernia, pES identified a VUS in LRP2, not reported by postmortem ES, and associated for one case with a RLIM variant (previously diagnosed). Discrepancies between prenatal and postmortem VUS are summarized in Table 2. Following our stepwise strategy, pES was negative for four fetuses with VUS identified by postmortem ES (Fig. 1).

From left to right, variant identified by postmortem ES (original FOETEX study) and match with variants identified by prenatal reanalysis.
Discussion
We report the first pilot study comparing the performance of ES for multisystemic CA before and after fetal autopsy.
In our series, trio-pES was completely concordant with postmortem ES for LP/P variant, either typical or atypical phenotype. Considering variability of fetal phenotype, trio-pES should be systematically favored. However, when parental DNA is not available, solo-pES could be an alternative with a highly concordance with postmortem ES (92%). Our data suggest that the prenatal phenotype, although limited, is not a limitation for interpretation of pES in case of multisystemic CA.
The implementation of pES in standard care would change the practice of prenatal diagnosis and require a highly specialized multidisciplinary team for prenatal imaging, interpretation of genetic variants and validation by phenotype-genotype correlation. During pregnancy, reverse-phenotyping using additional imaging or biological tests could be necessary to corroborate diagnosis in case of a LP/P variant or confirm, or not, causality of VUS, one major challenge of pES (4 to 32%) [12, 13]. Discrepancy between VUS considered by prenatal and postmortem analysis could be explained by poor phenotype-genotype correlations, limited in-silico prediction scores for selected variants, and clinical features included in nonspecific phenotype. Contrary to personalized postnatal medicine, there are no guidelines for reporting VUS from pES [11]. The uncertainty and limitations in prenatal counseling, and the large spectrum of adverse consequences must be considered carefully [9]. Therefore, we suggest to only considered VUS in OMIM genes with a high potential for clinical significance and if complementary tests are available to reclassify variant. Finally, for negative pES, reanalysis of ES using additional data provided by longitudinal pregnancy follow-up could lead to a diagnosis that was initially not considered. Currently, postnatal and postmortem clinical data of genetic disorders are mainly reported in clinical databases. The lack of specific database reporting prenatal data limits the development of the genomic prenatal medicine. It is now essential to develop them.
Implementation of pES and increase of prenatal data may influence indications of postmortem examinations. While fetal autopsy provides additional features in around 70% of cases, but it influences in only 25% of cases. In a large majority of cases, added value of fetal autopsy is limited with a total agreement with prenatal imaging or minor additional features in 45% and 30% of procedures respectively [14]. The implementation of whole-genome analyses should encourage us to reconsider the additive value of fetal autopsy. This is consistent with our results that show similar efficiency of trio-pES and postmortem ES. When pES identify LP/P variant in typical phenotypes, autopsy could be reconsidered because of no added value for genetic counseling. Conversely, postmortem examination remains essential in negative pES, VUS or atypical ultrasound phenotype secondary to a LP/P variant to look for additional phenotypic arguments and/or perform complementary etiological investigations on fetal tissues different from amniotic or fetal blood. New approach of fetal autopsy, based on virtual autopsy with or not or minimal invasive sampling, could be considered according to the prenatal phenotype [15]. These options should reduce parent’s reluctance and improve genetic counseling.
Previously offered after the pregnancy, implementation of pES may have a major clinical utility for patients and medical teams. Without diagnostic failure, our data suggest that offering pES does not limit the genetic counseling. However, the limits of ES (identification of structural, noncoding/epigenetics variations or repeat expansions) must be considered according to the fetal phenotype and common related disorders [16]. According to fetal phenotype, the timing of genetics tests offered should consider parental wishes following the pre-test counseling. pES also improves the prenatal prognostic assessment and help parents in decision-making about pregnancy outcome (termination or continuation of the pregnancy). It is also a key factor for medical teams about acceptance of late TOP in some countries or customization of perinatal care: adjustment of monitoring, planning delivery, and neonatal care (resuscitation, specific medications, palliative care…) [17]. pES appears as a powerful tool for personalized fetal medicine [18].
In conclusion, pES could now be extensively implemented in clinical practice to investigate MCA with a similar effectiveness than postmortem ES. A trio strategy should be favored. This study also leads to reconsider the practices of fetal autopsy when pES identify a causal etiological diagnosis.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author (Pr Christel THAUVIN-ROBINET, mail: christel.thauvin@chu-dijon.fr) on reasonable request.
References
Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, et al. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015;385:430–40.
Best KE, Rankin J, Dolk H, Loane M, Haeusler M, Nelen V, et al. Multilevel analyses of related public health indicators: the European Surveillance of Congenital Anomalies (EUROCAT) Public Health Indicators. Paediatr Perinat Epidemiol. 2020;34:122–9.
Hillman SC, McMullan DJ, Hall G, Togneri FS, James N, Maher EJ, et al. Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2013;41:610–20.
Normand EA, Braxton A, Nassef S, Ward PA, Vetrini F, He W, et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018;10:74.
Harris S, Gilmore K, Hardisty E, Lyerly AD, Vora NL. Ethical and counseling challenges in prenatal exome sequencing. Prenat Diagn. 2018;38:897–903.
Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Melbourne Genomics Health A, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. 2018;26:644–51.
Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747–57.
Pratt M, Garritty C, Thuku M, Esmaeilisaraji L, Hamel C, Hartley T, et al. Application of exome sequencing for prenatal diagnosis: a rapid scoping review. Genet Med. 2020;22:1925–34.
Wou K, Weitz T, McCormack C, Wynn J, Spiegel E, Giordano J, et al. Parental perceptions of prenatal whole exome sequencing (PPPWES) study. Prenat Diagn. 2018;38:801–11.
Lefebvre M, Bruel A-L, Tisserant E, Bourgon N, Duffourd Y, Collardeau-Frachon S, et al. Genotype-first in a cohort of 95 fetuses with multiple congenital abnormalities: when exome sequencing reveals unexpected fetal phenotype-genotype correlations. J Med Genet. 2021;58:400–13.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Deden C, Neveling K, Zafeiropopoulou D, Gilissen C, Pfundt R, Rinne T, et al. Rapid whole exome sequencing in pregnancies to identify the underlying genetic cause in fetuses with congenital anomalies detected by ultrasound imaging. Prenat Diagn. 2020;40:972–83.
Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758–67.
Godbole K, Bhide V, Nerune S, Kulkarni A, Moghe M, Kanade A. Role of fetal autopsy as a complementary tool to prenatal ultrasound. J Matern Fetal Neonatal Med. 2014;27:1688–92.
Cannie M, Votino C, Moerman P, Vanheste R, Segers V, Van Berkel K, et al. Acceptance, reliability and confidence of diagnosis of fetal and neonatal virtuopsy compared with conventional autopsy: a prospective study. Ultrasound Obstet Gynecol. 2012;39:659–65.
Burdick KJ, Cogan JD, Rives LC, Robertson AK, Koziura ME, Brokamp E, et al. Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am J Med Genet A. 2020;182:1400–6.
Tolusso LK, Hazelton P, Wong B, Swarr DT. Beyond diagnostic yield: prenatal exome sequencing results in maternal, neonatal, and familial clinical management changes. Genet Med. 2021;23:909–17.
Bianchi DW. From prenatal genomic diagnosis to fetal personalized medicine: progress and challenges. Nat Med. 2012;18:1041–51.
Acknowledgements
The authors thank the parents for their participation. The authors also thank cnrgh for exome sequencing, the University of Burgundy Center de Calcul (ccuB) for providing technical support and management of the informatics platform, and the genematcher platform for data sharing.
Funding
This work was funded by the Program Hospitalier de Recherche Clinique (PHRC) Interregional 14-013, the Regional Council of Burgundy and the “Fonds Européen de Développement Régional” (FEDER). Several authors are members of the ERN ITHACA. This study was funded by interregional French Phrc interregional 14-013 FOeTeX.
Author information
Authors and Affiliations
Contributions
Conceptualization: CT-R. Data curation: YD. Formal analysis: NB, ML, A-LB, PK, SN, CP, JT, FTM-T, SM, AS, AG, JD, AV. Funding acquisition: JT, CT-R, ML. Writing-original draft: NB. Writing-review and editing: CT-R.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All fetuses were initially included in this Foetex study, approved by our regional institutional review board and ethics committee (Comité de Protection des Personnes (CPP) EST I (Dijon)). Informed written consent was obtained from all subjects and participating family members.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bourgon, N., Garde, A., Bruel, AL. et al. Same performance of exome sequencing before and after fetal autopsy for congenital abnormalities: toward a paradigm shift in prenatal diagnosis?. Eur J Hum Genet 30, 967–975 (2022). https://doi.org/10.1038/s41431-022-01117-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01117-7
This article is cited by
-
Exome sequencing—one test to rule them all?
European Journal of Human Genetics (2022)
