
Abstract
Hereditary leiomyomatosis and renal cell carcinoma caused by loss-of-function germline variants of the FH gene can develop into aggressive renal cell carcinoma (RCC). We report the case of a 27-year-old man who died of RCC. Genetic testing revealed a novel pathogenic variant of FH, NM_000143.3:c.1013_1014del (p.Ile338Serfs*3), that was also identified in healthy siblings. Identification of genetic causes in the proband helped us to provide relatives with precise genetic counseling and appropriate surveillance programs.
Similar content being viewed by others
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a newly emerging hereditary tumor syndrome with an autosomal dominant inheritance pattern characterized by cutaneous leiomyoma, uterine leiomyoma, and renal cell carcinoma (RCC)1. More than 300 families with HLRCC have been reported to date, although the condition may be underdiagnosed2,3. Loss-of-function germline variants of the FH gene, which is located on chromosome 1q43 and encodes fumarate hydratase, cause HLRCC4. The incidence of RCC in FH pathogenic variant carriers is estimated to be approximately 15–20% with a median age of onset of 35–44 years3,5,6; however, a substantial number of FH pathogenic variant carriers can develop RCC at a very young age. Actually, a case of a seven-year-old boy with HLRCC-associated RCC was recently reported7. Histologically, these renal tumors are characterized by a unique type 2 papillary architecture but can also have a variety of patterns, such as cystic, tubular, solid, or mixed patterns8. HLRCC-associated RCC is typically aggressive and metastatic, and most patients with this type of RCC are at an advanced stage at the time of diagnosis. Thus, surveillance and early detection are of great importance9. Herein, we report a familial case of HLRCC caused by a novel pathogenic variant of FH.
A 26-year-old Japanese man was admitted to our hospital with right inguinodynia and motor dysfunction of the right lower limb. His medical history was unremarkable. Notably, his mother died of cancer of unknown primary origin at the age of 41 years, nine years before his admission. She had pelvic tumors with multiple metastatic lesions in the lungs, skin, left kidney, and vertebra. Malignant transformation of uterine leiomyomas was suspected, but an accurate assessment of the primary origin was not provided. The patient’s maternal grandfather died of pulmonary adenocarcinoma at the age of 74 years, and none of the other family members had HLRCC-associated episodes (Fig. 1a).

a The patient (III-1) died of renal cell carcinoma at the age of 27 years. His mother (II-2) died of cancer of unknown primary origin at age 41 years. The pathogenic variant of FH was identified in the patient as well as his two younger brothers (III-2 and III-3). P, proband; Roman numerals represent generation numbers; Arabic numerals indicate individual numbers; females are represented by circles and males by squares; E+ denotes the FH variant carrier; vertical bar denotes asymptomatic variant carrier. b Abdominal computed tomography revealed a solid mass in the left kidney (arrows) and an adjacent paravertebral tumor (arrowheads) of the patient.
On admission, the patient’s laboratory tests were normal, except for mildly elevated levels of C-reactive protein (0.69 mg/dL) and lactate dehydrogenase (617 U/L). He did not develop any cutaneous manifestations. Enhanced computed tomography scanning revealed a solid mass measuring approximately 60 mm in the left kidney and an adjacent paravertebral tumor (Fig. 1b), coupled with multiple pulmonary nodules and osteolytic lesions. Although pathological examination was not available at this point, RCC was suspected with TNM stage cT4NxM1. The patient was considered inoperable; thus, combination therapy with the immune checkpoint inhibitors nivolumab and ipilimumab was initiated. After the second administration, ipilimumab was intolerable due to the exacerbation of immune-related adverse effects. Subsequent treatment with nivolumab alone was continued but found to be ineffective. Thus, pazopanib and cabozantinib were administered with palliative radiation therapy.
Given the early onset of cancer with an aggressive phenotype in the patient as well as his mother, we proceeded to conduct genetic investigations as described in the Supplementary Methods after providing genetic counseling and obtaining written consent. Germline multigene panel testing identified a novel heterozygous frameshift variant NM_000143.3:c.1013_1014del (p.Ile338Serfs*3) in the FH gene in the patient, thereby confirming the diagnosis of HLRCC-associated RCC. A multiplex ligation-dependent probe amplification (MLPA) assay revealed no copy number changes in FH (data not shown).
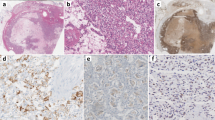
Thereafter, the tumor continued to progress, and the patient eventually died 13 months after the initiation of treatment. An autopsy confirmed an invasive substantial tumor measuring 45 × 25 × 15 mm at the upper part of the left kidney (Fig. 2a). Microscopically, the tumor exhibited a predominantly papillary growth pattern (Fig. 2b). At a higher magnification, clear and eosinophilic tumor cells possessed large nuclei with eosinophilic and enlarged nucleoli surrounded by a halo (Fig. 2c), which is the histological hallmark of HLRCC-associated RCC10. Immunohistochemistry for FH revealed minimal staining (Fig. 2d), demonstrating FH-deficient RCC. Additional Sanger sequencing analysis of the FH variant using blood- and tumor-derived DNA showed that the mutant allele was predominant in the tumor (Fig. 2e), suggesting loss of heterozygosity (LOH).

a Surface section of the left kidney. A yellowish-white solid tumor was observed. b Hematoxylin and eosin staining of the tumor demonstrated a predominantly papillary architectural pattern. c Higher magnification of the tumor showed that clear and eosinophilic tumor cells possessed large nuclei with eosinophilic and enlarged nucleoli surrounded by a halo. d Immunohistochemistry for FH was almost negative. e Schematic presentation of the FH gene (NM_000143.3) and pathogenic variant identified in this study. Sanger sequencing data of the FH gene confirmed a frameshift variant c.1013_1014del (p.Ile338Serfs*3) in the blood and tumor samples of the patient (highlighted by a gray box). Of note, the mutant allele was predominant in the tumor, indicating loss of heterozygosity.
Two younger siblings of the patient aged 22 and 23 years, who had no remarkable medical history, decided to undergo genetic testing after genetic counseling sessions. The results demonstrated that both had the same FH variants. At that time, no renal tumors or cutaneous manifestations were noted in the siblings. Hence, annual surveillance with magnetic resonance imaging for HLRCC-associated RCC and regular skin check-ups were scheduled.
The clinical course of the mother was reminiscent of HLRCC, although genetic testing had not been conducted. Given that two siblings shared the FH variant with the proband, the mother was assumed to harbor the same variant and be affected by HLRCC-associated malignancy. However, it is unclear whether the variant emerged de novo in the mother or was inherited from the maternal grandparents. In this regard, 20 Japanese families of HLRCC have been reported to date, and different FH variants have been reported in these families (Supplementary Table S1). The variants are presumed to be independent of each other; thus, a founder effect in the Japanese population is less likely.
The identified germline FH variant c.1013_1014del (p.Ile338Serfs*3) has hitherto not been reported. The variant is predicted to cause a frameshift in exon 7 of the FH gene, which can result in truncation of downstream amino acids. Of note, analysis of the somatic mutations of the tumor did not reveal any other pathogenic variants in the FH gene but revealed an LOH pattern. Specifically, the wild-type allele was lost, and the germline variant was retained. The occurrence of such an LOH event in the FH gene, which is frequently observed in HLRCC-associated RCC11, would be of substantial importance, thereby confirming that Knudson’s two-hit theory for tumor suppressor genes can be applied to FH. Of note, the minor existence of the wild-type allele in the tumor (Fig. 2e) was thought to be attributable to tumor heterogeneity or contamination of normal cells, including capillary endothelial cells within the dissected tumor sample.
In summary, we report a novel pathogenic variant of FH in a family with HLRCC. Identification of an underlying genetic cause in the patient helped us to provide family members with precise genetic counseling based on proper assessment of the genetic risk and appropriate surveillance programs in the future. Considering the documented childhood onset and aggressive phenotype of HLRCC-associated RCC, genetic testing and early intervention are crucial for at-risk family members of HLRCC.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3116.
References
Launonen, V. et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl Acad. Sci. USA 98, 3387–3392 (2001).
Alam, N. A. et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch. Dermatol. 141, 199–206 (2005).
Chayed, Z., Kristensen, L. K., Ousager, L. B., Rønlund, K. & Bygum, A. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J. Rare Dis. 16, 34 (2021).
Tomlinson, I. P. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 (2002).
Menko, F. H. et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam. Cancer 13, 637–644 (2014).
Hol, J. A. et al. Renal cell carcinoma in young FH mutation carriers: case series and review of the literature. Fam. Cancer 19, 55–63 (2020).
Taniguchi, R. et al. A patient with very early onset FH-deficient renal cell carcinoma diagnosed at age seven. Fam. Cancer https://doi.org/10.1007/s10689-021-00268-8 (2021).
Schmidt, L. S. & Linehan, W. M. Genetic predisposition to kidney cancer. Semin. Oncol. 43, 566–574 (2016).
Muller, M. et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin. Genet. 92, 606–615 (2017).
Merino, M. J., Torres-Cabala, C., Pinto, P. & Linehan, W. M. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am. J. Surg. Pathol. 31, 1578–1585 (2007).
Furuya, M. et al. Clinicopathological and molecular features of hereditary leiomyomatosis and renal cell cancer-associated renal cell carcinomas. J. Clin. Pathol. 73, 819–825 (2020).
Acknowledgements
The authors are grateful to the patient and family members for participating in this study. We also thank Dr. Shuji Mikami for his pathological assistance. This work was supported by grants from the Japan Society for the Promotion of Science (JSPS; 19H03628, 20K19082, and 21K19751), National Cancer Center Research and Development Fund (25-A-1 and 31-A-2), and Japan Agency for Medical Research and Development (AMED; JP19ck0106268 and JP20ck0106554).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yagi, Y., Abeto, N., Shiraishi, J. et al. A novel pathogenic variant of the FH gene in a family with hereditary leiomyomatosis and renal cell carcinoma. Hum Genome Var 9, 3 (2022). https://doi.org/10.1038/s41439-021-00180-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00180-8
This article is cited by
-
Fumarate hydratase (FH) and cancer: a paradigm of oncometabolism
British Journal of Cancer (2023)
-
Nivolumab
Reactions Weekly (2022)
