
Abstract
Rafflesia is a unique plant species existing as a single flower and produces the largest flower in the world. While Rafflesia buds take up to 21 months to develop, its flowers bloom and wither within about a week. In this study, transcriptome analysis was carried out to shed light on the molecular mechanism of senescence in Rafflesia. A total of 53.3 million high quality reads were obtained from two Rafflesia cantleyi flower developmental stages and assembled to generate 64,152 unigenes. Analysis of this dataset showed that 5,166 unigenes were differentially expressed, in which 1,073 unigenes were identified as genes involved in flower senescence. Results revealed that as the flowers progress to senescence, more genes related to flower senescence were significantly over-represented compared to those related to plant growth and development. Senescence of the R. cantleyi flower activates senescence-associated genes in the transcription activity (members of the transcription factor families MYB, bHLH, NAC, and WRKY), nutrient remobilization (autophagy-related protein and transporter genes), and redox regulation (CATALASE). Most of the senescence-related genes were found to be differentially regulated, perhaps for the fine-tuning of various responses in the senescing R. cantleyi flower. Additionally, pathway analysis showed the activation of genes such as ETHYLENE RECEPTOR, ETHYLENE-INSENSITIVE 2, ETHYLENE-INSENSITIVE 3, and ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR, indicating the possible involvement of the ethylene hormone response pathway in the regulation of R. cantleyi senescence. Our results provide a model of the molecular mechanism underlying R. cantleyi flower senescence, and contribute essential information towards further understanding the biology of the Rafflesiaceae family.
Similar content being viewed by others
Introduction
Rafflesia is a genus of holoparasitic angiosperm from the family Rafflesiaceae. Among its family members, Rafflesia is the most species-rich genus, comprising over 30 species with many new species being described in recent years1,2,3. Rafflesia is unique among other flowering plants due to its highly reduced vegetative structures, yet produces the largest flower in the world4. The largest flower recorded is Rafflesia arnoldii measuring up to 100 cm in diameter5 and the vegetative structure exists as endophyte made up of a uniseriate strand that transitions to flowering abruptly4. The floral structure comprised of congenitally fused petal whorl, which made up the diaphragm and five brick red perianth lobes, corresponding to sepals in most angiosperms6.
Rafflesia has a very long-life cycle that can take up to four years7, followed by a short blooming period of four to eight days8,9. Rafflesia cantleyi takes between six to 21 months for flower buds to mature and their blooms were recorded to last for only five to eight days10. During the blooming period, female Chrysomya chani carrion flies will be attracted to the flower, allowing pollination to happen11. Subsequently, the flower will start to die and early symptoms of death or aging (also known as senescence) in Rafflesia include withering and discoloration of perigone lobes from red to blackish-brown. Eventually, death can be seen when the entire flower decomposes.
Senescence is the last developmental stage of a plant’s life cycle, which involves highly coordinated structural, biochemical, and molecular changes that lead to the death of organs and the whole plant12. Senescence involves death at the cellular level known as programmed cell death (PCD)13 and is known to be triggered by various environmental factors such as heat and light stresses, insect and pathogen attacks, wounding, and salinity14. Studies of flower senescence at the molecular level have revealed genes involved in various cellular activities, including protein degradation (e.g., PRT22 in Sandersonia; RbCP1 in Rosa), nutrient remobilization (e.g., pDcCP1 in Dianthus; DAFSAG2 in Narcissus), and nucleic acid degradation (e.g., DSA6 in Hemerocallis; PhNUC1 in Petunia; DcNUC1 in Dianthus)15,16,17. Other than that, genes related to various phytohormone pathways such as ethylene biosynthesis (e.g., 1-AMINOCYCLOPROPANE-1-CARBOXYLATE (ACC) SYNTHASE (ACS) and ACC OXIDASE (ACO) in Dianthus), ethylene induction (e.g., ethylene receptors ETR1, ETR2, ERS1, and ERS2 in Arabidopsis; DcERS1, DcERS2, and DcETR1 in Dianthus) and ethylene signaling (e.g., CONSTITUTIVE TRIPLE RESPONSE 1 (CTR1) in Arabidopsis; RhCTR1 and RhCTR2 in Rosa) are also known to be regulated by flower senescence17. As in other plants, senescence in Rafflesia is an important event. This is more so as the plant exists almost entirely as a single flower, and as such, senescence of the single organ correlates directly with plant death.
Although the uniqueness of Rafflesia has always been a fascination, the biology of this plant remains poorly understood. Part of this is due to sampling difficulties and hence a lack of molecular studies on this species. Previously, we conducted a transcriptomic study on the R. cantleyi flower18 with more comprehensive transcriptome analyses involving multiple stages of R. cantleyi floral buds19,20. Parts of these data were instrumental towards the discovery of its ability to retain some common plastid-associated processes, albeit missing the photosynthetic components21. In this study, the next-generation sequencing technology was used as a tool to generate transcriptome data of two different flower stages of R. cantleyi, which are defined by the approximate duration from anthesis, i.e., one day (flower stage 1; F1) and three days (flower stage 2; F2) after full flower blooming. The transcriptome was analyzed to identify novel senescence-associated genes and their regulation in Rafflesia flower senescence. The expression levels of selected genes was validated using reverse transcription-quantitative polymerase chain reaction (RT-qPCR).
Results
Transcriptome assembly and functional annotation
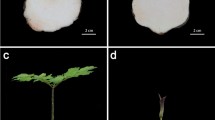
Transcriptome sequencing was carried out on the perigone lobe of the F1 and F2 flower stages of the R. cantleyi flower (Fig. 1). Rafflesia flowers have a short blooming period of up to seven days before they wither and die. From our observation in the field (data not shown), flowers after three days of blooming showed significant fungal infections, which will affect endogenous gene expression. Thus, to minimize this effect on the transcriptome analysis, we chose to focus on the F1 and F2 flower stages in this study.

The Rafflesia cantleyi flowers. R. cantleyi samples (a) one day (F1) and (b) three days (F2) after blooming were collected from Bukit Lahar, Pahang, Malaysia. Tissue samples from the perigone lobes of the two different flowers were obtained for transcriptome sequencing.
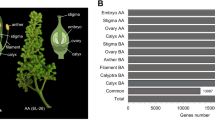
A total of 53.3 million cleaned reads from F1 and F2 were assembled using Trinity and iAssembler, generating 64,152 unigenes with an average length of 843 bp. BLASTX search resulted in a total of 39,234 (61%) and 26,775 (42%) unigenes matched to NCBI non-redundant (NR) and Swiss-Prot sequences, respectively. The similarity search performed on the unigenes against the Leaf Senescence Database (LSD 2.0) successfully identified 13,679 (21.3%) unigenes with significant matches to senescence-associated protein sequences (Supplementary Dataset S1). The BLAST analysis against LSD 2.0 (Fig. 2a) revealed that the majority of significant hits matches Arabidopsis thaliana (78.46%) and Musa acuminata (17.07%), followed by other plant species, such as Oryza sativa (2.19%), Zea mays (0.64%), and Glycine max (0.34%). Further classification of the unigenes into different categories according to their functions in senescence showed that the R. cantleyi unigenes are mostly involved in transcription regulation (1,287 unigenes), protein modification and degradation (1,206 unigenes), lipid and carbohydrate metabolism (880 unigenes), signal transduction (833 unigenes), and nutrient recycling (738 unigenes) (Fig. 2b).

Annotations of Rafflesia cantleyi transcriptome data. (a) Species distribution of annotated Rafflesia cantleyi flower unigenes based on LSD 2.0. (b) Functional categories of annotated Rafflesia cantleyi flower unigenes based on LSD 2.0.
Differential expression analysis and GO enrichment
Differential expression analysis was conducted to identify unigenes with significant differential expression between F1 and F2. The results of this analysis showed that a total of 5,166 unigenes identified were differentially expressed between F1 and F2. Of these, 4,062 unigenes were significantly up-regulated (log2FC ≥ 2; FDR ≤ 0.05) and 1,104 unigenes were significantly down-regulated (log2FC ≤ -2; FDR ≤ 0.05). Further analysis from LSD 2.0 showed that 1,073 unigenes were identified to be involved in senescence, of which 715 unigenes were up-regulated and 358 unigenes were down-regulated (Supplementary Dataset S2).
Gene ontology (GO) annotation carried out on the differentially expressed genes (DEGs) showed the highest match to cell (GO:0005623) and organelle (GO:0043226) for the cellular component category; catalytic activity (GO:0003824) and binding (GO:0005488) for the molecular function category; while the biological process category was dominated by metabolic (GO:0008152) and cellular process (GO:0009987) (Fig. 3). GO enrichment analysis of the DEGs was carried out to identify significantly over-represented DEGs (Supplementary Dataset S3). For up-regulated DEGs, 22 GO terms were significantly enriched, including transportation, lipid metabolism, cell cycle, response to stress, cellular protein modification process, nucleotide and protein binding, and enzyme regulator activity. In contrast, 27 GO terms for the down-regulated DEGs were significantly enriched, including meiotic nuclear division, response to biotic stimulus, post-embryonic development, and cell growth.

WEGO annotation plot of DEGs in Rafflesia cantleyi. The percentage and number of unigenes of each GO terms are indicated by the left and right x-axis, respectively. GO categories are indicated by the y-axis.
Other than that, the DEGs identified showed functions in the regulation of transcription factors, nutrient remobilization, redox regulation, and ethylene regulation (Table 1; Supplementary Dataset S4). Members of the transcription factor families MYB, bHLH, NAC, WRKY, bZIP, MADS-box, and AUX/IAA were found to be differentially expressed between the two flower stages. Furthermore, the expression of autophagy-related genes (ATG) and transporter genes involved in nutrient remobilization were up-regulated. A slight difference in the expression pattern was recorded for unigenes involved in redox regulation, in which most genes were constitutively expressed at both stages. This pattern of constitutive expression was similar to unigenes involved in the ethylene biosynthesis but unigenes involved in the ethylene signal transduction showed significant differential expression (Fig. 4).

KEGG pathway of ethylene biosynthesis and signal transduction pathway mapped with Rafflesia cantleyi unigenes. Green outlined boxes represent up-regulated unigenes while the red outlined box represents a down-regulated unigene. ER endoplasmic reticulum; ACO 1-AMINOCYCLOPROPANE-1-CARBOXYLATE OXIDASE; ACS 1-AMINOCYCLOPROPANE-1-CARBOXYLATE SYNTHASE; CTR1 CONSTITUTIVE TRIPLE RESPONSE 1; EBF1/2 EIN3-BINDING F-BOX PROTEIN; EIN2 ETHYLENE-INSENSITIVE 2; EIN3 ETHYLENE-INSENSITIVE 3; ERF1/2 ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR 1/2; ETR ETHYLENE RECEPTOR; MPK6 MITOGEN-ACTIVATED PROTEIN KINASE 6; SIMKK MITOGEN-ACTIVATED PROTEIN KINASE KINASE.
Expression analysis of selected DEGs by RT-qPCR
Seven genes with different expression patterns between the two flower stages were selected for RT-qPCR validation analysis. Results showed that QUINOLINATE SYNTHASE (QS) has the highest increase in expression, followed by ETHYLENE RECEPTOR (ETR), ATG4, and CATALASE (CAT) (Fig. 5a), while PECTIN ESTERASE (PE) has the highest decrease in expression, followed by MYB and AGL8 (Fig. 5b). Overall, the RT-qPCR results showed a high correlation with that of RNA-seq (R2 > 0.8742) (Fig. 5c).

RT-qPCR analysis on the fold change of expression level at F2 relative to expression level at F1. (a) Unigenes with up-regulated expression. (b) Unigenes with down-regulated expression. (c) Correlation plot between the RT-qPCR fold-change (FC) compared to FC calculated from TPM values of RNA-seq analysis. Genes studied are CAT CATALASE, ATG4 AUTOPHAGY-RELATED, ETR ETHYLENE RECEPTOR, QS QUINOLINATE SYNTHASE, AGL8 AGAMOUS-LIKE 8, MYB MYELOBLASTOSIS, and PE PECTIN ESTERASE. Asterisks indicate significant differences (P < 0.05) in gene expressions.
Discussion
In this study, besides the more commonly used databases such as NR and Swiss-Prot, a BLAST search was also performed against the leaf senescence-specific LSD 2.0 database. In the absence of a specific database for flower senescence, the LSD 2.0 database was utilized since most of the leaf senescence genes can also be found in the flower. Furthermore, leaf and floral organs arise from the same primordium, thus it is expected that the senescence genes are conserved in these two organs, as observed in A. thaliana22 and Erysimum linifolium23. BLAST results showed that the majority of the homologs are from A. thaliana (79.14%) and M. acuminata (16.98%), and this may be partly due to both of these species having the largest datasets in the LSD 2.0 database, 69.89% for A. thaliana and 16.46% for M. acuminata. GO annotation revealed that DEGs were mainly responsible for fundamental biological regulation and metabolism common in other plants. This suggests that R. cantleyi flower senescence is a coordinated biological process that could be regulated similarly to other plants24.
The transcriptional control mechanism, which involves various transcription factors, plays a pivotal role in regulating gene expression during plant development and senescence25. Many transcription factors have been identified previously and found to be differentially expressed during the development and senescence of various flower systems17. In R. cantleyi, members of MYB, bHLH, NAC, and WRKY were found to be up-regulated at F2. All of these families were known to be involved in various kinds of biological processes, including responses to stresses and injuries26,27,28,29. MYB102 functions in response to biotic and abiotic stresses, such as defense against insects, injuries, and osmotic stresses30,31,32, whereas, MYB7 is a transcription factor involved in the flavonoid biosynthesis induced during salinity stress33. On the other hand, the function of bHLH family members MYC234, bHLH09227, and ICE135,36,37 in abiotic stress response has been well characterized. Moreover, expression data from genome-wide transcriptome analyses in many plants such as A. thaliana38, soybean39, rice40, and Chrysanthemum lavandulifolium41 revealed that a significant proportion of NAC genes are related to stress responses42. Studies conducted on the expression of WRKY showed rapid induction when plants are exposed to a variety of stresses or defensive signals, including plant defense against attack and senescence29,43,44.
The up-regulation of these transcription factors is in line with other studies on flower senescence. The same phenomenon can be seen in Dianthus and Gardenia, which showed a high level of expression in MYB during flower senescence45,46. Other than that, the expression pattern of bHLH between the two R. cantleyi flower stages is similar to that in Gardenia, whereby this transcription factor family plays a major role in regulating flower senescence46,47. The expression pattern of NAC in the R. cantleyi flower was similar to that of A. thaliana leaf and flower senescence22,48.
The transcription factors that showed significant down-regulation in R. cantleyi were identified to be from the bZIP, MADS-box, and AUX/IAA families, which play important roles in flower development49,50,51. The transcription factors from the bZIP family plays important roles in regulating seed maturation and flower development49. These functions are also shared with the MADS-box transcription factors, which are known to be involved in the development of the reproductive structure and determination of meristems and organ identity50. Additionally, AUX/IAA transcription factors are also involved in many aspects of plant development. IAA1, IAA3, and IAA4 were identified to play key roles in development, particularly in root morphology and gravitropism, stem elongation, and inflorescence formation in A. thaliana, O. sativa, and Eucalyptus51,52,53. The down-regulation of these transcription factors suggests their reduced function during flower senescence. Overall, transcription factors involved in various responses to stresses, injuries, and senescence were up-regulated contrary to the down-regulation of transcription factors involved in flower development.
Flowers have a fixed lifespan determined by their status in sexual reproduction such that flowers that have been pollinated or are no longer receptive to pollination will be programmed to senensce54. In some plant species, the symptoms of flower senescence are visibly shown through wilting or withering of petals, whereas in other plants, the petals abscise while still turgid55. For plant species with petal wilting, such as in Rafflesia, the main function of flower senescence is proposed to allow the remobilization of nutrients to other parts of the plant16,56,57. One of the processes during the remobilization of nutrients is protein degradation, which involves multiple protease enzymes. These enzymes degrade proteins by hydrolyzing internal peptide bonds, or more commonly known as proteolysis, and this activity is irreversible.
Cysteine protease is an enzyme that has been reported to be involved in the remobilization of essential nutrients from senescing floral tissue. In some plant species such as Sandersonia58, Narcissus59, Alstroemeria56, and Rosa hybrida60, genes encoding cysteine protease were found to be induced during flower senescence. In this study, a unigene encoding cysteine protease in the R. cantleyi flower was identified. Differential expression analysis showed that the cysteine protease gene ATG4 (UN020537) was up-regulated at F2 (Table 1; Supplementary Dataset S4). This gene was identified to be involved in autophagy, which is the main pathway for the degradation of proteins in vacuole activated by environmental stresses61.
Other than that, unigenes encoding other related autophagy proteins were also identified. Besides ATG4, the up-regulation of ATG7, ATG8, and ATG9 at F2 were also similar to those in A. thaliana during leaf senescence62. The expression patterns of ATG4 and ATG8 in R. cantleyi are similar to Ipomea nil and Petunia with increasing expression during petal senescence63,64,65. Furthermore, the activation of the autophagy pathway is negatively regulated by TARGET OF RAPAMYCIN (TOR). The down-regulation of TOR caused the autophagy pathway activation in A. thaliana66. In this study, a unigene that encodes TOR was down-regulated at F2, indicating that the autophagy mechanism in the R. cantleyi flower is probably activated by the regulation of TOR.
Besides that, unigenes involved in transporter activity, such as ABC transporter and transporters of sugar, nitrates, and polyamines were also identified as DEGs (Table 1; Supplementary Dataset S4). In A. thaliana, the expression of transporter protein was found to be up-regulated during leaf senescence, which reflects nutrient remobilization during senescence67. Overall, the differential expression of unigenes related to nutrient remobilization suggests transportation of nutrients from the senescing flower of R. cantleyi, perhaps for the development of seeds and fruits.
The reactive oxygen species (ROS) represent free radicals derived from oxygen molecules, such as singlet oxygen (1O2), superoxide (O2·−), hydroxyl (OH·), hydroperoxyl (HO2·−), and hydrogen peroxide (H2O2)68. These radicals can oxidize various molecules in plants, which gives adverse effects on membrane integrity and protein stability leading to programmed cell death69. ROS are produced during normal plant development and the production increases when triggered by environmental stresses such as drought, salinity, and pathogen attack. ROS are extremely harmful in high concentrations, thus they need to be removed from the cell through various activities of antioxidative enzymes, such as superoxide dismutase (SOD), catalase (CAT), and ascorbate oxidase (ASO). In previous studies of flower senescence, such as in Gladious70, Hemerocallis71,72, and Rosa hybrida73, various free oxygen radicals were detected in flower petals. The increase of these free radicals caused the up-regulation of antioxidative enzymes SOD, CAT, and ASO during flower senescence55. In this study, unigenes that are involved in redox regulation encoding antioxidative enzymes CAT (UN004263), SOD (UN012078), ASO (UN037470), and other peroxidases (UN001999, UN063210) were identified (Table 1; Supplementary Dataset S4). Our results revealed that these unigenes showed constitutive expressions at both flower stages, except CAT (UN004263), which was found to be up-regulated in F2. High expression of ROS-related unigenes at both flower stages indicates the regulation of redox activity in R. cantleyi.
Flower senescence involves highly coordinated events that are regulated by endogenous signals, such as phytohormones and environmental factors, including biotic and abiotic stresses74. Several plant hormones have been reported to be involved in senescence, of which ethylene and abscisic acid (ABA) function to induce flower senescence, while cytokinin, gibberellic acid (GA), and auxin delay the process16,75. In most plant species, ethylene is a phytohormone that initiates flower senescence76. Results from differential expression analysis were able to help identify genes involved in ethylene hormone regulation in R. cantleyi.
Ethylene is synthesized through cysteine and methionine metabolism pathways mediated by enzyme precursors ACO and ACS77. In this study, unigenes encoding ACO (UN064036) and ACS (UN023149) were constitutively expressed at both flower stages. However, unigenes involved in the ethylene signal transduction pathway were identified to be differentially expressed. Among them, positive regulators ETHYLENE-INSENSITIVE 2 (EIN2) (UN007860), ERF (UN012030), and ETR (UN024806), and transcription factor ETHYLENE-INSENSITIVE 3 (EIN3) (UN025327) were significantly up-regulated, whereas a negative regulator CTR1 (UN004001) was significantly down-regulated at F2 (Table 1; Supplementary Dataset S4). ETR serves as a receptor that binds to ethylene gases and the loss of ETR function delays senescence in flowers and leaves as recorded in Petunia and Nicotiana sylvestris78,79,80. On the other hand, in the ethylene signal transduction pathway, the binding of ethylene to the ETR receptor causes the CTR gene to be inactive. The deactivation of CTR activates other downstream components such as transcription factors EIN2, EIN3, and ERF, followed by ethylene response17. Constitutive expression of unigenes involved in the ethylene biosynthesis and the up-regulation of unigenes involved in the ethylene signal transduction pathway suggest the role of ethylene regulation in flower senescence of R. cantleyi (Fig. 4).
Results of the transcriptome data analysis allowed us to build a model for the regulation of senescence in R. cantleyi, based on a senescence regulation pathway proposed by Tripathi and Tuteja (2007)81 (Fig. 6). Senescence involves death at the cellular level known as PCD13. In R. cantleyi, PCD might be induced by environmental factors, such as biotic and abiotic stresses, which hasten the process of flower senescence and lead to the regulation of ethylene hormone and various transcription factors. Various pathways involving phytohormones and the regulation of transcription and signal transduction are known to activate the expression of senescence-associated genes17. The regulation of these transcription factors suggests activation of senescence-associated genes that are involved in the process of nutrient remobilization and redox regulation.

A model of flower senescence regulation in Rafflesia cantleyi. Upward arrows represent up-regulated genes and downward arrows represent down-regulated genes. Genes in parentheses are constitutively expressed. ASO L-ASCORBATE OXIDASE; ATG AUTOPHAGY-RELATED GENES; CAT CATALASE; CTR CONSTITUTIVE TRIPLE RESPONSE; EIN ETHYLENE-INSENSITIVE; ERF ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR; ETR ETHYLENE RECEPTOR; SOD SUPEROXIDE DISMUTASE; TOR TARGET OF RAPAMYCIN.
Conclusion
Transcriptome generation and analysis focused on DEGs for two stages of R. cantleyi flowers yielded a model on the regulation of flower senescence in R. cantleyi. This model consists of the regulation of transcription factors and ethylene-related genes, together with nutrient remobilization, redox regulation, and the activation of senescence-associated genes, which ultimately lead to a coordinated death of a floral organ. The understanding of key genes and important pathways involved in R. cantleyi flower senescence will contribute towards further studies, including in the fields of biodiversity and conservation of this plant family.
Methods
Sample preparation and transcriptome sequencing
R. cantleyi flowers were collected from Bukit Lahar, Pahang, Malaysia, (Permission from the Pahang State Forestry Department—reference no. PHN. PHG. (PEM) 118/146 Bhg. 3 (123)). The collection of plant material complied with relevant regulations. Flower samples were obtained from two different stages of flower development, i.e., one day (flower stage 1; F1) and three days (flower stage 2; F2) after blooming (Fig. 1). Tissue samples were acquired from the perigone lobes of two different flowers that were collected from the same area of sampling and under a relatively similar environment. These samples were dissected and surface-sterilized using 10% (v/v) Clorox and rinsed with distilled water on site. Following that, the tissue samples were flash-frozen in liquid nitrogen before being transported to the laboratory. All the samples collected were stored at − 80 °C until further use. Total RNA extraction was carried out based on the modified CTAB method82. The extracted RNA was quantified using the ND-1000 Nanodrop spectrophotometer (Thermo Fisher Scientific Inc., USA). Before sequencing, the RNA integrity was quantified using the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). cDNA libraries were constructed with the TruSeq® Stranded Total RNA Library Prep Kit (Illumina, USA) according to the manufacturer’s protocol. Subsequently, the libraries were sequenced using Illumina HiSeq™ 2000.
Data pre-processing and de novo assembly of transcriptome
Raw sequencing reads generated (deposited to the NCBI SRA database with the accession numbers SRR7544088 (F1) and SRR7544087 (F2)) were quality-filtered using SolexaQA (v.2.2)83. High-quality reads were acquired by removing adaptor sequences and low-quality reads with Phred score less than 20 and read length less than 50 bp. Furthermore, the reads were screened for contaminating sequences from other organisms by aligning them using Bowtie2 (v2.3.0)84 to all genomes of bacteria (version 66, 7 July 2014), viruses, and fungi (version 73, 2 Nov 2015) from the NCBI database. Reads that aligned to these genomes were compared against the plant database Plaza 3.085 and reads without any alignment to the plant genomes were discarded. Finally, the paired-end clean reads were subjected to de novo assembly using Trinity (version v2.2.0)86 and iAssembler (version v1.3.2)87. The R. cantleyi flower transcriptome assembly was deposited in the TSA database with the accession number GIQT00000000.
Functional annotation of R. cantleyi transcriptome
To predict the putative functions of assembled unigenes, functional annotation was performed using BLASTX against NR and Swiss-Prot protein databases with an E-value cutoff of 1E-6. Unigenes involved in senescence were identified by performing similarity searches against 44 species of dicots and monocots in the Leaf Senescence Database (LSD 2.0)88. Gene ontology (GO) annotation was implemented on the annotated sequences from NR using the Blast2GO program and was further classified into three categories: biological process, molecular function, and cell component. The summary of GO classification was visualized using WEGO89. GO enrichment analysis was performed using Fisher’s exact test (FDR < 0.05) based on the up-regulated and down-regulated unigenes to predict the overrepresented GO function. KEGG pathway assignment was performed on unigenes using KEGG Automatic Annotation Server (KAAS)90 to predict metabolic pathways involved in the R. cantleyi flower.
Differential gene expression analysis
Gene expression levels were estimated by mapping paired-end clean reads from each flower stage (F1 and F2) to the assembled transcriptome using the RSEM method91. Due to the great difficulties of on-site sampling of specific Rafflesia flower stages in the rainforest and the inability of cultivating Rafflesia for study under laboratory or controlled environment conditions, only one biological replicate was available for transcriptomic analysis. The gene expression was based on TPM (transcripts per kilobase million) that normalizes transcript length first before sequencing depth. Unigenes with TPM values greater than 0 were considered as expressed. By pairwise comparisons of the two libraries (F1 vs. F2), the DEGs were identified using the edgeR package92 based on the read counts of unigenes in different libraries. Trimmed Mean of M-values (TMM) normalization with an automatic method to estimate dispersion without replicates allowed single-replicate DEG analysis in edgeR, assuming that the counts are not too small. The significance of differential gene expression was judged by the false discovery rate (FDR ≤ 0.05) and fold change (|Log2FC|≥ 2) as the cut-off threshold.
Gene expression analysis by RT-qPCR
Seven DEGs related to transcription factors, nutrient remobilization, redox regulation, and ethylene hormone pathway were selected from the transcriptome dataset and expression profiles were investigated by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis. Gene-specific primers (Supplementary Dataset S5) were designed using Primer-Blast. TUBULIN (TUB) and ELONGATED FACTOR 1A (EF1A) were selected as reference genes after they were identified as the most stable genes in the R. cantleyi flower by the Bestkeeper software93. For RT-qPCR analysis, 20 ng/µL DNAse-treated RNA was used as a template in a 20 µL total reaction with QuantiNova® SYBR® Green RT-PCR Kit (Qiagen, USA) according to the manufacturer’s instructions. The reactions were incubated in the CFX96™ Real-Time Detection System (Bio-Rad, USA) with the following cycling conditions: 10 min at 50 °C and 2 min at 95 °C for the reverse transcription, 40 cycles of 5 s at 95 °C and 10 s at 60 °C. In this analysis, three perigone lobes of each flower stage were treated as biological replicates, and each PCR reaction included three technical replicates. The changes in gene expression were calculated based on the 2−∆∆Ct method94. Statistical analysis of the data was performed using unpaired Student’s t-test at a P < 0.05 significance level. Correlation analysis of relative expression values from RNA-seq and RT-qPCR was performed using Microsoft Excel.
References
Galindon, J. M. M., Ong, P. S. & Fernando, E. S. Rafflesia consueloae (Rafflesiaceae), the smallest among giants; a new species from Luzon Island, Philippines. PhytoKeys, 37 (2016).
Jumaat, H. A. et al. Rafflesia tuanku-halimii (Rafflesiaceae), a new species from Peninsular Malaysia. Sains Malaysiana 45, 1589–1595 (2016).
Susatya, A., Hidayati, S. N. & Riki, S. Rafflesia kemumu (Rafflesiaceae), a new species from Northern Bengkulu, Sumatra, Indonesia. Phytotaxa 326, 211–220 (2017).
Nikolov, L. A. et al. Holoparasitic Rafflesiaceae possess the most reduced endophytes and yet give rise to the world’s largest flowers. Ann. Bot. 114, 233–242 (2014).
Barkman, T. J. et al. Accelerated rates of floral evolution at the upper size limit for flowers. Curr. Biol. 18, 1508–1513 (2008).
Nikolov, L. A. et al. Developmental origins of the world’s largest flowers, Rafflesiaceae. Proc. Natl. Acad. Sci. USA 110, 18578–18583 (2013).
Meijer, W. Rafflesiaceae. Flora Malesiana 13, 1–42 (1997).
Hidayati, S. N., Meijer, W., Baskin, J. M. & Walck, J. L. A contribution to the life history of the rare Indonesian holoparasite Rafflesia patma (Rafflesiaceae). Biotropica 32, 408–414 (2000).
Meijer, W. A contribution to the taxonomy and biology of Rafflesia arnoldii in West Sumatra. Annales Bogoriensis 3, 33–44 (1958).
Nais, J. Rafflesia of the World. (Natural History Piblications, Kota Kinabalu, Malaysia, 2001).
Wee, S. L., Tan, S. B. & Jürgens, A. Pollinator specialization in the enigmatic Rafflesia cantleyi: a true carrion flower with species-specific and sex-biased blow fly pollinators. Phytochemistry 153, 120–128 (2018).
Ahmad, S. S. & Tahir, I. How and why of flower senescence: understanding from models to ornamentals. Indian J. Plant Physiol. 21, 1–11 (2016).
Danon, A., Delorme, V., Mailhac, N. & Gallois, P. Plant programmed cell death: a common way to die. Plant Physiol. Biochem. 38, 647–655 (2000).
Velasco-Arroyo, B. et al. Senescence-Associated Genes in Response to Abiotic/Biotic Stresses. in Progress in Botany Vol. 79 (eds. Cánovas, F., Lüttge, U. & Matyssek, R.) 89–109 (Springer, Cham, 2017).
Dar, R. A., Tahir, I. & Ahmad, S. S. Physiological and biochemical changes associated with flower development and senescence in Dianthus chinensis L. Indian J. Plant Physiol. 19, 215–221 (2014).
Rogers, H. J. From models to ornamentals: How is flower senescence regulated?. Plant Mol. Biol. 82, 563–574 (2013).
Shahri, W. & Tahir, I. Flower senescence: Some molecular aspects. Planta 239, 277–297 (2014).
Lee, X.-W. et al. Perigone lobe transcriptome analysis provides insights into Rafflesia cantleyi flower development. PLoS ONE 11, e0167958 (2016).
Amini, S. et al. RNA-seq data from different developmental stages of Rafflesia cantleyi floral buds. Genom. Data 14, 5–6 (2017).
Amini, S. et al. Transcriptome landscape of Rafflesia cantleyi floral buds reveals insights into the roles of transcription factors and phytohormones in flower development. PLoS ONE 14, e0226338 (2019).
Ng, S.-M. et al. Comparative analysis of nucleus-encoded plastid-targeting proteins in Rafflesia cantleyi against photosynthetic and non-photosynthetic representatives reveals orthologous systems with potentially divergent functions. Sci. Rep. 8, 1–10 (2018).
Wagstaff, C., Yang, T. J., Stead, A. D., Buchanan-Wollaston, V. & Roberts, J. A. A molecular and structural characterization of senescing Arabidopsis siliques and comparison of transcriptional profiles with senescing petals and leaves. Plant J. 57, 690–705 (2009).
Price, A. M. et al. A comparison of leaf and petal senescence in wallflower reveals common and distinct patterns of gene expression and physiology. Plant Physiol. 147, 1898–1912 (2008).
Ülker, B., Shahid Mukhtar, M. & Somssich, I. E. The WRKY70 transcription factor of Arabidopsis influences both the plant senescence and defense signaling pathways. Planta 226, 125–137 (2007).
Balazadeh, S., Riaño-Pachón, D. & Mueller-Roeber, B. Transcription factors regulating leaf senescence in Arabidopsis thaliana. Plant Biol. 10, 63–75 (2008).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Jiang, Y., Yang, B. & Deyholos, M. K. Functional characterization of the Arabidopsis bHLH92 transcription factor in abiotic stress. Mol. Genet. Genomics 282, 503–516 (2009).
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K. & Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms 1819, 97–103 (2012).
Jiang, J. et al. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 59, 86–101 (2017).
De Vos, M. et al. The Arabidopsis thaliana transcription factor AtMYB102 functions in defense against the insect herbivore Pieris rapae. Plant Signal. Behav. 1, 305–311 (2006).
Ding, Y. et al. Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 13, 229. https://doi.org/10.1186/1471-2229-13-229 (2013).
Persak, H. & Pitzschke, A. Dominant repression by Arabidopsis transcription factor MYB44 causes oxidative damage and hypersensitivity to abiotic stress. Int. J. Mol. Sci. 15, 2517 (2014).
Mondal, S. K. & Roy, S. Genome-wide sequential, evolutionary, organizational and expression analyses of phenylpropanoid biosynthesis associated MYB domain transcription factors in Arabidopsis. J. Biomol. Struct. Dyn. 33, 1577–1601 (2018).
Abe, H. et al. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15, 63–78 (2003).
Chinnusamy, V. et al. ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 17, 1043–1054 (2003).
Gilmour, S. J., Sebolt, A. M., Salazar, M. P., Everard, J. D. & Thomashow, M. F. Overexpression of the Arabidopsis CBF3 transcriptional activator mimics multiple biochemical changes associated with cold acclimation. Plant Physiol. 124, 1854–1865 (2000).
Lee, B.-H., Henderson, D. A. & Zhu, J.-K. The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 17, 3155–3175 (2005).
Jiang, Y. & Deyholos, M. K. Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 6, 1–20 (2006).
Le, D. T. et al. Genome-wide survey and expression analysis of the plant-specific NAC transcription factor family in soybean during development and dehydration stress. DNA Res. 18, 263–276 (2011).
Fang, Y., You, J., Xie, K., Xie, W. & Xiong, L. Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol. Genet. Genom. 280, 547–563 (2008).
Huang, H. et al. Transcriptome-wide survey and expression analysis of stress-responsive NAC genes in Chrysanthemum lavandulifolium. Plant Sci. 193, 18–27 (2012).
Shao, H., Wang, H. & Tang, X. NAC transcription factors in plant multiple abiotic stress responses: progress and prospects. Front. Plant Sci. 6, 902 (2015).
Song, Y., Ai, C.-R., Jing, S.-J. & Yu, D.-Q. Research progress on functional analysis of rice WRKY genes. Rice Sci. 17, 60–72 (2010).
Bakshi, M. & Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 9, e27700 (2014).
Hoeberichts, F. A., Van Doorn, W. G., Vorst, O., Hall, R. D. & Van Wordragen, M. F. Sucrose prevents up-regulation of senescence-associated genes in carnation petals. J. Exp. Bot. 58, 2873–2885 (2007).
Tsanakas, G. F., Manioudaki, M. E., Economou, A. S. & Kalaitzis, P. D. novo transcriptome analysis of petal senescence in Gardenia jasminoides Ellis. BMC Genom. 15, 554 (2014).
Trivellini, A., Cocetta, G., Hunter, D. A., Vernieri, P. & Ferrante, A. Spatial and temporal transcriptome changes occurring during flower opening and senescence of the ephemeral hibiscus flower Hibiscus rosa-sinensis. J. Exp. Bot. 67, 5919–5931 (2016).
Balazadeh, S. et al. A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J. 62, 250–264 (2010).
Jakoby, M. et al. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 7, 106–111 (2002).
Theissen, G. et al. A short history of MADS-box genes in plants. Plant Mol. Biol. 42, 115–149 (2000).
Uberti-Manassero, N. G., Lucero, L. E., Viola, I. L., Vegetti, A. C. & Gonzalez, D. H. The class I protein AtTCP15 modulates plant development through a pathway that overlaps with the one affected by CIN-like TCP proteins. J. Exp. Bot. 63, 809–823 (2012).
Song, Y. & Xu, Z.-F. Ectopic overexpression of an auxin/indole-3-acetic acid (Aux/IAA) gene OsIAA4 in rice induces morphological changes and reduces responsiveness to auxin. Int. J. Mol. Sci. 14, 13645–13656 (2013).
Yu, H. et al. Comprehensive genome-wide analysis of the Aux/IAA gene family in Eucalyptus: Evidence for the role of EgrIAA4 in wood formation. Plant Cell Physiol. 56, 700–714 (2015).
Jones, M. L. Mineral nutrient remobilization during corolla senescence in ethylene-sensitive and -insensitive flowers. AoB PLANTS 5, plt023-plt023 (2013).
van Doorn, W. G. & Woltering, E. J. Physiology and molecular biology of petal senescence. J. Exp. Bot. 59, 453–480 (2008).
Wagstaff, C. et al. Cysteine protease gene expression and proteolytic activity during senescence of Alstroemeria petals. J. Exp. Bot. 53, 233–240 (2002).
Rogers, H. & Munné-Bosch, S. Production and scavenging of reactive oxygen species and redox signaling during leaf and flower senescence: Similar but different. Plant Physiol. 171, 1560–1568 (2016).
Eason, J. R., Ryan, D. J., Pinkney, T. T. & O’Donoghue, E. M. Programmed cell death during flower senescence: isolation and characterization of cysteine proteinases from Sandersonia aurantiaca. Funct. Plant Biol. 29, 1055–1064 (2002).
Hunter, D. A., Steele, B. C. & Reid, M. S. Identification of genes associated with perianth senescence in daffodil (Narcissus pseudonarcissus L.‘Dutch Master’). Plant Sci. 163, 13–21 (2002).
Hajizadeh, H. et al. Expression of genes encoding protein kinases during flower opening in two cut rose cultivars with different longevity. Iran. J. Biotechnol. 9, 230–233 (2011).
Wang, W., Xu, M., Wang, G. & Galili, G. Autophagy: An Important biological process that protects plants from stressful environments. Front. Plant Sci. 7, 2030 (2016).
Breeze, E. et al. High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 23, 873–894 (2011).
Yamada, T., Ichimura, K., Kanekatsu, M. & van Doorn, W. G. Homologs of genes associated with programmed cell death in animal cells are differentially expressed during senescence of Ipomoea nil petals. Plant Cell Physiol. 50, 610–625 (2009).
Shibuya, K., Shimizu, K., Yamada, T. & Ichimura, K. Expression of autophagy-associated ATG8 genes during petal senescence in Japanese morning glory. J. Jpn. Soc. Hortic. Sci. 80, 89–95 (2011).
Shibuya, K., Niki, T. & Ichimura, K. Pollination induces autophagy in petunia petals via ethylene. J. Exp. Bot. 64, 1111–1120 (2013).
Liu, Y. & Bassham, D. C. TOR Is a negative regulator of autophagy in Arabidopsis thaliana. PLoS ONE 5, e11883 (2010).
Buchanan-Wollaston, V. et al. Comparative transcriptome analysis reveals significant differences in gene expression and signalling pathways between developmental and dark/starvation-induced senescence in Arabidopsis. Plant J. 42, 567–585 (2005).
Rogers, H. J. Is there an important role for reactive oxygen species and redox regulation during floral senescence?. Plant Cell Environ. 35, 217–233 (2012).
De Pinto, M., Locato, V. & De Gara, L. Redox regulation in plant programmed cell death. Plant Cell Environ. 35, 234–244 (2012).
Fobel, M., Lynch, D. V. & Thompson, J. E. Membrane deterioration in senescing carnation flowers coordinated effects of phospholipid degradation and the action of membranous lipoxygenase. Plant Physiol. 85, 204–211 (1987).
Panavas, T. & Rubinstein, B. Oxidative events during programmed cell death of daylily (Hemerocallis hybrid) petals. Plant Sci. 133, 125–138 (1998).
Chakrabarty, D., Verma, A. K. & Datta, S. K. Oxidative stress and antioxidant activity as the basis of senescence in Hemerocallis (day lily) flowers. J. Hortic. For. 1, 113–119 (2009).
Fukuchi-Mizutani, M. et al. Molecular and functional characterization of a rose lipoxygenase cDNA related to flower senescence. Plant Sci. 160, 129–137 (2000).
Iqbal, N. et al. Ethylene role in plant growth, development and senescence: Interaction with other phytohormones. Front. Plant Sci. 8, 475 (2017).
Gan, S. Annual Plant Reviews: Senescence Processes in Plants. Vol. 26 (John Wiley & Sons, 2008).
Reid, M. S. & Wu, M.-J. Ethylene and flower senescence. Plant Growth Regul. 11, 37–43 (1992).
Wang, K.L.-C., Li, H. & Ecker, J. R. Ethylene biosynthesis and signaling networks. Plant Cell 14, S131–S151 (2002).
Rodrıguez, F. I. et al. A copper cofactor for the ethylene receptor ETR1 from Arabidopsis. Science 283, 996–998 (1999).
Wilkinson, J. Q. et al. A dominant mutant receptor from Arabidopsis confers ethylene insensitivity in heterologous plants. Nat. Biotechnol. 15, 444–447 (1997).
Yang, T. F., Gonzalez-Carranza, Z. H., Maunders, M. J. & Roberts, J. A. Ethylene and the regulation of senescence processes in transgenic Nicotiana sylvestris plants. Ann. Bot. 101, 301–310 (2008).
Tripathi, S. K. & Tuteja, N. Integrated signaling in flower senescence. Plant Signal. Behav. 2, 437–445 (2007).
Abdul-Rahman, A. et al. RNA extractions of mangosteen (Garcinia mangostana L.) pericarps for sequencing. Sains Malaysiana 46, 1231–1240 (2017).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinf. 11, 485 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357 (2012).
Proost, S. et al. PLAZA 3.0: An access point for plant comparative genomics. Nucleic Acids Res. 43, D974–D981 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Zheng, Y., Zhao, L., Gao, J. & Fei, Z. iAssembler: A package for de novo assembly of Roche-454/Sanger transcriptome sequences. BMC Bioinformatics 12, 453 (2011).
Li, Z. et al. LSD 2.0: An update of the leaf senescence database. Nucleic Acids Res. 42, D1200–D1205 (2014).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297 (2006).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 1–16 (2011).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotech. Lett. 26, 509–515 (2004).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
The work was supported financially by Research University Grants from Universiti Kebangsaan Malaysia (Grant No. GUP-2016-008 and LAUREATE-2013-001).
Author information
Authors and Affiliations
Contributions
N.A.M.E., H.H.G. and K.L.W. conceived and designed the experiments. H.A., M.N.M.I., M.A.A.J. and J.H.A. collected the samples. H.A., M.F.A.B. and M.N.M.I. participated in the data generation and transcriptome assembly. N.A.M.E. performed the data analysis. N.M.I., H.H.G. and K.L.W. contributed to the project management and monitoring. N.A.M.E., K.R., H.H.G. and K.L.W. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohd-Elias, NA., Rosli, K., Alias, H. et al. Transcriptome analysis of Rafflesia cantleyi flower stages reveals insights into the regulation of senescence. Sci Rep 11, 23661 (2021). https://doi.org/10.1038/s41598-021-03028-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03028-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
