
Abstract
Cancers harboring mutations in the Kirsten rat sarcoma homolog (KRAS) gene have been associated with poor prognosis and lack of targeted therapies. KRAS mutations occur in approximately one in four patients diagnosed with non-small cell lung cancer (NSCLC) with KRAS G12C mutations harbored at approximately 11–16%. Research into KRAS-driven tumors and analytical chemistry have borne a new class of selective small molecules against the KRAS G12C isoform. Phase II data for sotorasib (AMG510) has demonstrated a 37.1% overall response rate (ORR). Adagrasib (MRTX849) has demonstrated a 45% ORR in an early study. While single agent efficacy has been seen, initial data suggest combination approaches are an opportunity to improve outcomes. Here, we present perspectives on the initial progress in targeting KRAS G12C, examine co-mutations evident in KRAS G12C NSCLC, and comment on potential future combinatorial approaches including SHP2, SOS1, MEK, EGFR, mTOR, CDK, and checkpoint blockade which are currently being evaluated in clinical trials. As of May 28, 2021, sotorasib has achieved US FDA approval for patients with KRAS G12C mutant lung cancer after one line of a prior therapy.
Similar content being viewed by others

Mechanism of KRAS oncogenesis and chemistry
The KRAS protein oscillates between the inactive form of GDP-RAS and the active form of GTP-RAS1,2,3. The protein includes two major binding regions including the G domain and the C terminal membrane targeting region which, once activated through guanine nucleotide exchange factors (GEFs), have the ability to further affect many pathways including PI3K, mTOR, RAF/MEK/ERK, and other downstream cell cycle pathways4. To cause the shift in state of GTP-bound RAS into GDP-bound RAS, GTPase activating proteins (GAPs) hydrolyze GTP Fig. 1 leading to a reduction in cell growth seen when GTP is bound to RAS and serving as a functional control mechanism of proliferation5. Somatic mutations block the reversible nature of this gene, creating a constitutively bound RAS-GTP complex and resulting conformational change.

RAS/MAPK pathway noting therapeutic targets and some associated therapies currently being evaluated.
G12C mutations make up about 13% of NSCLC, 3–5% of colorectal cancers, and 1–2% of other cancer types. A genomic analysis of KRAS mutations in NSCLC showed that a G>A transition has been increasingly seen in those who have never smoked, while a G>T transversion is commonly seen in current and former smokers who are diagnosed with lung cancer6. In comparison, KRAS mutations are less common in lung squamous cell carcinoma (<1–5%) and small cell lung cancer (1–2%)7,8,9,10. Among other types of cancers, KRAS mutations appear in approximately 97% of pancreatic adenocarcinomas, 52% of colorectal cancers, and some responses have been seen in these tumor types albeit with varying consistency3. Within a subgroup analysis of those who are more frequently diagnosed with KRAS G12C NSCLC, both Caucasian and African American populations can be more often diagnosed than Asian populations. Interestingly, white females harbor KRAS G12C NSCLC mutations more often than their male counterparts. This trend is opposite for Asian populations, as Asian males more frequently have NSCLC G12C mutations11.
Cancers harboring RAS mutations have remained genomically untargetable more than 30 years after the initial discovery of the oncogene, and attempts to target KRAS directly or through downstream mechanisms have been met with limited efficacy in clinical trials. Earlier attempts to target KRAS have focused on molecules that bind directly to RAS, inhibit interaction with its activator SOS or its effector RAF, affect downstream signaling (i.e., MEK inhibitors), act on synthetically lethal pathways, and affect binding at the plasma membrane (i.e., farnesyltransferase inhibitors). Unfortunately, none of these mechanisms have resulted in approvals for these strategies in KRAS G12C mutant cancers. KRAS binds tightly with GTP (with picomolar affinity in vivo) in the main binding pocket, and its features sterically inhibit many treatments from reversing the strong binding affinity for GTP4. In order for RAS to maintain its functionality, the molecule requires a modification with C15 farnesyltransferase when localized in the plasma membrane3. This modification was the target of an active pipeline of farnesyltransferase inhibitors and yielded preclinical responses however limited responses in clinical trials12.
We have modeled KRAS G12C and presented areas of binding of the medicine sotorasib at the allosteric pocket under the Switch II loop region (Fig. 2) in the GDP-bound state of KRAS using ChimeraX software from UCSF13,14. Binding at this pocket has been found to inhibit RAS activity by blocking SOS-mediated nucleotide exchange and altering the affinity of KRAS for GDP versus GTP nucleotide. These innovative approaches demonstrated the presence of previously unknown binding pockets on the surface of RAS and provided a framework for continued efforts to develop novel RAS targeted therapies15,16. Small molecules bind irreversibly with high specificity to the mutant cysteine at exon 12, leading to preferential GDP binding over GTP16. Decreased binding affinity to the downstream RAF protein target contributed anti-tumor effects. One of the first developed mutant specific, covalent small molecule inhibitors to demonstrate proficiency in targeting KRAS G12C and locking it into a GDP-bound state was ARS-853 by Wellspring Biosciences and had robust cellular activity against KRAS G12C in the low micromolar range. Modifications were made to this small molecule inhibitor, giving rise to ARS-1620, which targeted the His95 amino acid on KRAS G12C. ARS-1620 also proved to have a higher potency and preclinical efficacy and produced a better conformational response than its predecessor1.

A General representation of KRAS G12C protein in 3-dimensional space in relationship with bound sotorasib and GDP. The outline box represents area that is focused on in (B) view. B Distances between localized KRAS G12C side-chains to sotorasib (in pink, encircled in white) are presented. Switch pockets 1 and 2 are highlighted in dark green and red, respectively. Loci we identified included: Val9, Lys16, Gln61, Glu63, Arg68, His95, Tyr96, and Gln99. This has been modelled via UCSF ChimeraX13,14.
In vitro and pre-clinical trial data demonstrates anti-tumor activity
Direct inhibition of KRAS G12C was validated by ARS-1620, but the identification of improved inhibitors suitable for clinical testing proved difficult. A key challenge was suboptimal potency owing to the small volume of the pocket occupied by ARS-1620, which offered limited avenues for additional protein-ligand interactions. The discovery that a surface groove, created by an alternative orientation of His95, could be occupied by aromatic rings and enhance interactions with the KRAS G12C protein was important to identify therapeutic vulnerabilities (Fig. 2). Sotorasib (AMG 510) emerged as the top candidate from optimization of His95 groove-binding molecules. Although portions of the AMG 510 and ARS-1620 ligands are structurally related, the His95 groove binding is a novel feature of AMG 510. Enhanced interactions improved the potency of AMG 510 as compared to ARS-1620 in a nucleotide-exchange assay with recombinant GDP-bound KRAS G12C. The kinetics of the reaction between AMG 510 and GDP-KRAS G12C were measured by mass spectrometry and exhibited a marked improvement compared to ARS-1620. Preclinical work in a variety of cancer cell lines from lung cancer and other solid tumors harboring the G12C mutation demonstrated anti-tumor response17.
In parallel, a structure-based drug design approach and optimization led to the discovery of adagrasib (MRTX849) as a potent, covalent KRAS G12C inhibitor. In xenografts and preclinical models, a precursor tool compound MRTX1257 showed promising results, leading to development of MRTX84918,19. The preclinical maximally efficacious dosage that promoted optimal tumor regression while maintaining tolerance was between 30-100 mg/kg per day. Pre-clinical mouse models (CDX and PDX) also showed a total response rate of 65% across tumors, and specifically a response rate of 75% in NSCLC models20. In pre-clinical models utilizing H358 and MIAPaCa-2 xenografts, evaluation of adagrasib showed that concomitant mutations (i.e., TP53, KEAP1, or STK11) did not predict against therapeutic response.
The function of KRAS and its RAF-MEK-ERK pathway depends largely on the activation of the KRAS scaffold, comprised of proteins including Src homology region 2 domain-containing phosphatase 2 (SHP2) and son of sevenless homolog 1 (SOS1). The combined interactions of SHP2 and SOS1 transition the GDP-bound state of KRAS to the GTP-bound state, rendering the protein active and allowing for proliferation. SHP2 and SOS1 have been demonstrated to serve as drug targets in controlling a mutant KRAS. In addition, SHP2 activation enables resistance mechanisms to MEK inhibitor therapy and enhances expression of signaling upstream from KRAS21. SOS1 has been labeled as a potential oncogenic driver, with the capability of tumorigenesis in vivo and the ability to activate both the RAS protein through a RAS-GEF (guanine nucleotide exchange factor) and Myc target genes, as well as increase phosphorylation of MEK22. This provided the rationale for a therapeutic approach of combining a KRAS G12C inhibitor with a MEK inhibitor to anticipate the potential resistance, and early investigations with AMG510 demonstrated preclinical efficacy. In another study evaluating SOS1 interactions in the KRAS pathway, chemical inhibition of SOS1 with BAY-293 completely suppressed the pathway in wild-type KRAS cells. In mutant-KRAS tumor cells, inhibition of SOS1 led to a 50% reduction in phospho-ERK. When combined with the KRAS G12C inhibitor, ARS-853 and BAY-293 worked together in NCI-H358 cells to produce a synergistic response23. Additional in vitro combination screen assays were performed to determine the possible combination therapies that may further support adagrasib and sotorasib combinatorial strategies. Possible escape mechanisms for the tumor were identified through in vitro and in vivo models and CRISPR screens and included HER family receptor tyrosine kinases, EGFR, SHP2, and mTOR/S6 pathways19.
Scaffold proteins can mediate therapeutic resistance mechanisms to reactivate the RAS/MAPK pathway by RTK signaling. KRAS G12C inhibitors have been characterized by adaptive resistance through re-activation of KRAS scaffold proteins, emphasizing the importance of targeting RTKs and other cooperative resistance mechanisms in potential combinations. KRAS G12C inhibition via ARS-1620 has been shown to cause temporary pathway suppression followed by RAS-MAPK rebound activation in KRAS G12C mutant lung cell lines. KRAS G12C inhibition in combination with the RTK inhibitors afatinib and erlotinib have demonstrated synergy in vitro models. The use of combination SHP2 and KRAS G12C inhibition have been shown to decrease tumor growth in in vivo models compared to monotherapy alone. SHP2 has been described to be a mediator between RTK signaling and KRAS activation. SHP2 inhibition combined with KRAS G12C inhibition decreased KRAS-GTP active conformation and MAPK pathway activation. The described benefit of SHP2 and KRAS G12C inhibition supports further investigation of combination therapy approach to prevent upregulation of upstream pathways24.
Based on non-small cell lung cancer datasets within cBioPortal, the landscape of KRAS G12C mutated tumors includes a complex landscape of co-altered genes (Fig. 3)25,26. Frequently co-altered genes include TP53, STK11, KEAP1, and CDKN2A. The significance of targeting the co-altered genes is being explored in ongoing studies and biomarkers beyond genomic aberrations include RNA-based signatures and protein expression.

Clinical trials show initial efficacy
Sotorasib
The FDA approval of sotorasib has been a remarkable example of the speed of recent FDA approvals for clinically active medicines. The phase I investigation of sotorasib was initiated in August 2018 with the CodeBreak 100 study (NCT #03600883). Preliminary results for the phase I study presented in 2019 showed a partial response (PR) for sotorasib in 5 of 10 patients with KRAS G12C-mutant NSCLC and 4 patients achieved stable disease (SD) for 90% disease control rate by investigator review. In 18 patients with KRAS G12C mutant colorectal cancer (CRC), 13 achieved SD27. Approximately 66.1% of patients reported adverse events (AEs) from the treatment, which included nausea and diarrhea. 18.6% of the total enrolled patients reported one or more AEs of grade of 3 or higher that were deemed treatment related. The mean elimination half-life of the drug was determined to be 5.5 h. Dose escalation ranged from 180 mg to 960 mg, and responses were seen in all dose levels evaluated.
In 2020, updated results of 59 evaluable NSCLC patients were presented and 19/59 achieved PR (32.2% ORR) and 33/59 achieved SD (88.1% DCR). Of the 59 patients, 34 were treated at 960 mg with an ORR of 35.3% and DCR of 91.2%. In colorectal cancer, 3 of 42 patients achieved PR (ORR 7.1%) and another 28/42 achieved SD (DCR 73.8%). 28 patients with diagnosed cancers other than NSCLC or colorectal cancer were evaluated, with four reaching PR and 17/28 achieving SD. Within the NSCLC cohort, the median progression-free survival (PFS) was 6.3 months, and within the colorectal cancer cohort the PFS was 4.0 months28.
In a more mature Phase II cohort presented in 2021, 126 patients with previously treated NSCLC were enrolled and presented, 124 of which were response eligible by central review. Results demonstrated an ORR of 37.1%, including 43 partial responses and 3 complete responses. Disease control rate was 80.6%29. Median duration of response was 10.0 months and median progression-free survival was 6.8 months. This data was granted FDA priority review on February 16, 2021 and the phase 3 clinical study comparing the efficacy of sotorasib versus docetaxel for patients with KRAS G12C-mutant NSCLC is actively ongoing30.
An updated analysis of the 126 patient Phase II NSCLC cohort has been recently published31. In the updated analysis, the overall response rate remained at 37.1% although one of the partial responses converted to a complete response. The updated analysis provided the first report of overall survival for NSCLC patients treated with a selective KRAS G12C inhibitor. The median overall survival was estimated at 12.5 months, which compares favorably to historical benchmarks in this disease setting (i.e. docetaxel combined with ramucirumab had 10.5 months overall survival in the REVEL study)32. Subgroup analyses demonstrated consistency of benefit for both overall response and overall survival across various subgroups including prior lines of therapy and prior treatment history33. Phase II and Phase III development proceeded with the 960 mg dose, although the sponsor has announced they will conduct a randomized study of sotorasib 240 mg versus sotorasib 960 mg as part of a US FDA post-marketing requirement34.
Adagrasib
In KRYSTAL-01 (NCT #03785249), a phase I/Ib and II trial (n = 79) to investigate the efficacy of adagrasib, the majority of patients were included in the dosage cohort of 600 mg taken twice daily, although doses up to 1200 mg twice daily were evaluated. In the combined Phase I/Ib and Phase II NSCLC cohorts of patients (n = 51) receiving 600 mg twice daily, 45% achieved PR while 51% achieved SD for a DCR of 96%. In the combined cohort of 18 patients with colorectal cancer who received 600 mg twice daily (n = 18), 17% achieved PR, while 78% achieved SD for a total disease control rate of 94%. In patients with other types of tumors (non-NSCLC, non-CRC) who were administered 600 mg twice daily (n = 6), ORR was determined to be 67% with one response unconfirmed and all patients remaining on treatment at data cutoff. The half-life of adagrasib was determined to be ~24 h. Combining all enrolled patients for safety analyses, 85% of patients enrolled in the clinical trial experienced AEs, with 30% experiencing grades 3–4, and 2% experiencing grade 5 adverse events. In the cases of fatal events, one patient experienced cardiac failure and another experienced pneumonitis35.
Exploratory analyses for both sotorasib and adagrasib have analyzed the impact of co-occurring alterations on efficacy. For sotorasib, an ORR of 50% was observed in 22 KRAS G12C patients with STK11 mutation and no KEAP1 mutation29. However, in the 13 patients with triple mutation of KRAS G12C, STK11, KEAP1, the ORR was 23%. For adagrasib, an ORR of 64% was observed in 14 KRAS G12C/STK11 co-altered patients (versus 33% in 30 KRAS G12C mutant/STK11 wild-type patients)35. The analysis of KEAP1 within the adagrasib STK11 subset has not been described at this time. The efficacy signals observed with STK11 co-alterations may provide insight into opportunities for evaluation in front-line NSCLC or with combination approaches. KRAS G12C inhibition may be a favorable approach in patients with concomitant STK11 mutations given findings that these patients may represent a population whom respond poorly to PD-1 therapy based on retrospective studies36.
Other molecules under investigation
Several other novel compounds are currently under active evaluation to understand differences in terms of potency and on target resistance (Table 1)28,37,38,39,40,41,42,43,44,45. A modification to the ARS-1620 molecule has been brought forward as JNJ-74699147 and entered clinical development in July 2019 by Janssen. The phase I study completed recruitment (NCT #04006301) and stopped enrollment with additional information not publicly disclosed at this time. Another KRAS G12C small molecule inhibitor, LY3499446, has been advanced by Eli Lilly but since discontinued and a new small molecule inhibitor with more selectivity is currently being evaluated as LY353798246.
Genentech is evaluating a KRAS G12C small molecule inhibitor GDC-6036 (NCT #04449874). This small molecule inhibitor has been shown to have high potency and selectivity47. InventisBio has developed a KRAS G12C inhibitor (D-1553), which has been positioned in NSCLC and colorectal cancer. The phase I/II is ongoing and will include evaluation of the drug as monotherapy and in combination with standard cytotoxic therapy (NCT #04585035)48. In the beginning of January 2021, Novartis KRAS G12C inhibitor compound JDQ443 is undergoing a phase Ib/II clinical trial as a monotherapy and in combination with either TNO155 or spartalizumab (NCT #04699188)49. Revolution Medicine is developing RMC-6236, a pan RAS inhibitor which may be able to target both exon 12 mutations and exon 13 mutations4.
In addition to the KRAS G12C molecules currently in clinical development, there are novel molecules in preclinical stage development that have additional differentiating characteristics. While all of the current clinical stage molecules bind to the inactive GDP-bound conformation of KRAS G12C, Revolution Medicines is advancing a compound, RMC-6291, that binds to the active GTP-bound conformation of KRAS G12C50. The compound binds to the active GTP-bound conformation via a tri-complex approach in which the molecule binds a chaperone protein and in turn complexes with the active KRAS conformation with expected IND filing in 202251. Meanwhile, others including Eli Lilly are developing more potent forms of GDP-bound KRAS G12C inhibition52. LY3537982 is a novel KRAS G12C inhibitor that binds KRAS G12C with higher potency and target occupancy than sotorasib and adagrasib in preclinical studies and has a first-in-human trial planned for 2021.
Pathways forward with combinatorial approaches
Clinical utility in previously untreated NSCLC and in other cancer types such as colorectal cancer will likely require greater efficacy than that seen to-date with KRAS G12C inhibitor monotherapy. Strong emphasis towards combination development is currently underway with a wide variety of combinatorial approaches currently under investigation including MEK, KRAS scaffold (SHP2, SOS1), EGFR, CDK4/6, mTOR, and cytotoxic chemotherapy (Table 1). The mechanisms described here are adapative signaling patterns that have been described preclinically after sotorasib or adagrasib exposure in tumor models (Fig. 1).
MEK inhibition
In vitro combinations have been evaluated combining sotorasib and inhibitors of HER kinase, EGFR, SHP2, PI3K/AKT, and MEK. Investigations evaluating both single agent and combinatorial efficacy of MEK inhibitors selumetinib and trametinib have demonstrated improvements in some clinical endpoints and have demonstrated responses across KRAS isoforms. Co-occurring STK11 and KRAS mutations in mouse models show increased resistance to MEK inhibition, compared to responses in mice with concomitant KRAS and p53 mutations. The addition of a MEK inhibitor to docetaxel demonstrated improved outcome in patients with KRAS-mutant NSCLC compared to either single agent treatment, combination with erlotinib, or docetaxel alone53. Based on the in vitro data, MEK inhibitors with lower dosages of sotorasib demonstrated an investigative opportunity forward4. Combinatorial efficacy of a MEK inhibitor with chemotherapy, as well as with the addition of a G12C inhibitor are being evaluated54. Clinical combinations of mTOR inhibitors with MEK inhibitors yielded severe adverse effects, and have prompted preclinical trials to test the substitution of a MEK inhibitor with the KRAS G12C inhibitor ARS-1620, in combination with an mTOR inhibitor (everolimus) and an insulin-like growth factor 1 receptor (IGF-1R) inhibitor (linsitinib)55. Both sotorasib and adagrasib are being evaluated in early clinical studies in combination with the MEK inhibitor trametinib.
KRAS Scaffold: SHP2 and SOS1
Other targets in the KRAS scaffold, namely SOS1 and SHP2, are also being actively investigated for combination potential. SHP2 (PTPN11) is a tyrosine phosphatase in which assists in activation of RAS through the RAS/MAPK pathway (Fig. 1). SHP2 inhibition can be efficacious in targeting for BRAF RAS-GTP cancers, inactivation of neurofibromin 1 (NF1), and RAS cancers in which an oscillation of RAS is seen. Through both knockout and inhibition of SHP2, SOS1 can be inhibited and RAS/MAPK is restricted in oncogenic proliferation56.
Multiple SHP2 inhibitors are being investigated in both monotherapy and combination with KRAS G12C inhibitors. Revolution Medicine and Sanofi have initiated development of RMC-4630, which is being investigated in phase I/II clinical trials as a monotherapy and in combination. Pre-clinical models have shown efficacy of RMC-4630 in combination with sotorasib57. A tool SHP2 inhibitor compound developed by Revolution Medicine is RMC-4550, which showed significant in vivo results when targeting KRAS G12C H358 and MIAPaCa-2 xenograft models both in terms of potency and sensitivity56. Relay Therapeutics has developed RLY-1971, an oral SHP2 inhibitor, and is investigating the drug as a monotherapy in a phase I clinical trial58. Genentech has collaborated on the development of RLY-1971 for a combination approach with KRAS G12C. Novartis is currently investigating the efficacy of TNO155 as monotherapy and in combination with G12C inhibition. Jacobio and AbbVie are jointly developing two oral small molecule SHP2 inhibitors including JAB-3068 and JAB-3312 which are being evaluated as monotherapies and in combination with a KRAS G12C inhibitor59.
A novel investigation into KRAS G12C therapeutic approaches has yielded a new target of the SOS1 catalytic domain that affects the EGFR/MAPK pathway. Through inhibition of SOS1, the binding of KRAS to GTP and the constitutively active state is reduced. Multiple KRAS G12 and G13 mutations are able to be inhibited. The SOS1:KRAS inhibitor BI-3046, developed by Boehringer Ingelheim, has been evaluated in vivo in NSCLC and CRC PDX models specifically in combination with trametinib (MEK inhibitor). Pre-clinical models showed that the combination of the SOS1:KRAS and MEK inhibition resulted in tumor regression. Thus far, the combination of trametinib with Boehringer Ingelheim’s SOS1:KRAS inhibitor BI 1701963 has shown both time- and dose-dependencies within MAPK pathway targets. Phase I monotherapy and combination therapy clinical trial results are expected soon, as BI 1701963 will be evaluated with both trametinib and adagrasib60.
EGFR, CDK4/6, mTOR
Sotorasib and adagrasib are being evaluated with CDK inhibitors, an mTOR inhibitor, MEK inhibitors, and EGFR inhibitors61. When achieving maximal response from adagrasib, association was seen with ERK activity inhibition. A HER family inhibitor (afatinib), CDK4/6 inhibitor (palbociclib), and mTOR pathway inhibitor may complement the efficacy of adagrasib with further improvements in response based on adaptive signaling. Afatinib, RMC-4550, vistusertib (mTOR), everolimus (mTOR), and palbociclib combinations were evaluated in cell line and xenograft models to explore dual pathway inhibition. Each tested combination proved to significantly enhance anti-tumor activity of adagrasib compared to any individual therapy alone19.
The role of CDK4/6 inibitors in KRAS mutated disease has been an active area of interest. Abemaciclib, a cyclin-dependent kinase (CDK) 4/6 inhibitor was investigated in the JUNIPER clinical trials against erlotinib in patients with KRAS mutant (exon 12 or exon 13) advanced NSCLC. The outcome showed a similar median OS between the two therapies, which was statistically insignificant. The cohort treated with abemaciclib experienced a PFS of 3.6 months, an ORR of 8.9%, and a DCR of 54.4%. The cohort treated with erlotinib experienced lower values, including a PFS of 1.9 months, an ORR of 2.7%, and a DCR of 31.7%. These latter three values were statistically significant, demonstrating clinical activity of CDK4/6 inhibition within KRAS exon 12 and exon 13 NSCLC patients62. Ongoing work evaluates CDK4/6 inhibitors in combination with G12C selective inhibitors19.
Immune checkpoint inhibitors in G12C NSCLC
Approved immunotherapies for NSCLC and/or SCLC are pembrolizumab, nivolumab, ipilimumab, atezolizumab, and durvalumab. PD-1 monoclonal antibody therapy has been approved as a first-line option for patients as either a monotherapy or in conjunction with platinum-based chemotherapy in NSCLC. Patients with KRAS G12C aberrations were included in frontline immunotherapy clinical trials including KEYNOTE-189, KEYNOTE-042, KEYNOTE-024, and CheckMate-227. Upon stratification of patients with KRAS G12C, it has been found in KEYNOTE-042 that the ORR for pembrolizumab monotherapy was 66.7%, while the ORR for chemotherapy was 23.5%. The median PFS for patients treated with pembrolizumab was 15 months, as compared to 6 months for patients treated with chemotherapy63. In patients with a concomitant mutation of STK11, however, the therapies had less benefit, leading to STK11 being labeled as a potentially negative predictor for immunotherapy however additional work is ongoing in this field36. In preclinical studies, KRAS G12C direct inhibitors were noted to upregulate a pro-inflammatory tumor microenvironment and enhance anti-tumor T cell activity. Enhancement of the immune activity led to response seen with sotorasib in CT-26 cell line models17. Currently, multiple trials are investigating the potential of G12C inhibitors with existing immune checkpoint inhibitors (Table 1). As anti-PD1 antibodies can have durable responses, the synergistic effects of the therapies may be important in patients with KRAS G12C-mutant NSCLC.
While clinical data for PD-1 checkpoint inhibitors and KRAS G12C combinations is highly anticipated, clinical data on the sequencing of PD-1/PD-L1 checkpoint inhibitors and sotorasib is an important area for exploration. Per recent subgroup analyses of the Phase II NSCLC cohort from CodeBreaK 100, patients appear to derive benefit after PD1/PD-L1 checkpoint inhibition, including the application of the checkpoint inhibitor within 3 months of sotorasib initiation31. The vast majority of patients (115 of 126) received prior PD-1/PD-L1 checkpoint inhibition and the response rate was 36.3% (vs. 37.1% in the overall population) and median overall survival was 12.0 months (versus 12.5 months in the overall population). Patients whom have had checkpoint therapy within 3 months had overall response of 34.4% and median overall survival of 11.7 months, compared to 39.7% and not evaluable in patients that had more than 3 months between checkpoint inhibition and sotorasib. These data may indicate that prior immune therapy does not meaningfully negatively impact the activity of sotorasib, suggesting a possible combination approach may be further explored given non-overlapping mechanisms of action.
Acquired resistance mechanisms
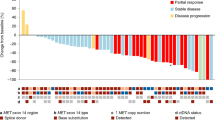
Mechanisms of resistance from direct G12C inhibitors are currently under investigation. A study recruited repeat biopsy or ctDNA samples from 23 patients with NSCLC and seven with CRC, all treated with adagrasib64. 42% of this cohort experienced multiple resistance mutations following treatment, while the rest of patients experienced a single resistance mutation. Mutations identified in the switch II binding pocket of KRAS included R68S, H95D, H95R, and Y96C. We observed in our structural model the proximity of Val9, Lys16, Gln61, Glu63, Arg68, His95, Tyr96, and Gln99 to bound sotorasib (Fig. 2). Given the position of these amino acids, mutations may impede binding or function of sotorasib. Alterations also occurred at the cysteine residue of G12, as well as in exon 13. Patients further acquired on-target amplifications of KRAS G12C and off-target alterations of BRAF, NRAS, MEK, and gene fusions. Importantly, a histological change was observed in some patients through a proposed transformation of adenocarcinoma to squamous cell carcinoma (SCC), without any further acquired resistance mechanisms64. Similar histological transformations have been observed after EGFR inhibitor therapy65. An understanding of how one G12C inhibitor compares to another based on mechanisms of resistance with mutations in the switch II binding pocket may be important in determining the sequence of therapy and treatment of resistance.
Future directions with G12C inhibitors
Is there front-line potential in NSCLC?
The most important alternative for G12C selective inhibitors in therapeutic landscape remain anti-PD1 therapy in patients with lung cancer. Immunotherapy with checkpoint inhibitors remain a standard of care for front line management in KRAS mutated NSCLC. Comparing the emerging KRAS G12C clinical data in NSCLC with chemotherapy and immunotherapy in KEYNOTE-189, with the caveat that patient characteristics differ between trials, activity of the KRAS G12C inhibitors in delaying disease progression appears more robust than standard platinum doublet therapy (PFS 4.9 months) however less than pembrolizumab (PD-1 inhibitor) plus platinum doublet therapy (PFS 9.0 months). KRAS mutation-positive patients derived benefit in both PFS and OS from the combination of pembrolizumab plus chemotherapy as opposed to platinum doublet therapy alone as first-line therapy for patients with metastatic NSCLC66. Sotorasib is being evaluated in combination with platinum chemotherapy and in combination with PD-1 inhibitors. Data suggesting that patients with KRAS-mutant NSCLC with STK11/LKB1 co-occurring mutation may have higher responses and may have attenuated responses to immune checkpoint therapy36. A cohort of the adagrasib KRYSTAL-01 study is evaluating adagrasib in previously untreated KRAS G12C/STK11 co-mutated NSCLC as this disease subset has poor prognosis with chemotherapy or immunotherapy and is an area of unmet need.
Do these molecules have dose-dependent CNS efficacy?
The central nervous system is an important space for targeted therapy drug development. It is important to note that patients with active brain metastases were excluded from both initial trials for both sotorasib and adagrasib, providing a lack of data on how the small molecule inhibitors penetrate the central nervous system. The sotorasib CodeBreak 101 has recently included an arm to specifically investigate the efficacy of monotherapy on patients with brain metastases61. Preclinical data of adagrasib efficacy on KRAS G12C brain metastases in a LU99Luc mouse model show a dose-dependent CNS effect with tumor regression noted20. Further work is necessary to confirm whether these drugs may have a neuroprotective effect67. As each of the inclusion criteria for both sotorasib and adagrasib did not permit patients with active brain metastases to participate, further studies are needed to assess CNS response.
Is there an opportunity to expand the reach of molecular profiling for mutation detection?
A major challenge in navigating the most effective therapeutic path for patients involves access to molecular profiling to identify which patients may have the presence of driver and resistance mutations, as approximately 10-25% of NSCLC cases are not eligible for a biopsy or do not have adequate tissue68,69. Next-generation sequencing of circulating tumor DNA in plasma samples using ultra-sensitive sequencing technologies with a short turn-around time have facilitated increased detection70. Concordance of tissue mutations detected in plasma samples can be ~90% in late-stage lung cancer71,72. Serial monitoring for the re-emergence of tumor-derived mutations is being evaluated to understand progression of disease based on radiographic imaging73. The current clinical trials have broadened eligibility to include circulating tumor DNA for some KRAS G12C trials. Amgen and Guardant have announced plans to develop a liquid biopsy companion diagnostic for sotorasib74.
Conclusion
The FDA has granted priority review for sotorasib, leading to approval in May of 2021 and availability of this compound for wide clinical use. Recent overall survival data has been favorable with an overall survival of 12.5 months including second and third line and beyond patients receiving benefit. Favorable ORR, DOR, PFS, and a favorable toxicity profile relative to current standards in the previously treated setting demonstrate high potential utility in clinical care. Ongoing work to identify the mechanisms of resistance to sotorasib and adagrasib can lead to rationale combinations of targeted therapy with second site mutations in KRAS identified as well as bypass pathways. Specifically how mechanisms of resistance to each compound may influence sequence of therapy and combinatorial strategies is an active area of ongoing exploration.
References
Janes, M. R. et al. Targeting KRAS mutant cancers with a covalent G12C-Specific Inhibitor. Cell 172, 578–589 (2018).
Arbour, K. C. et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin. Cancer Res. 24, 334–340 (2018).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014).
Nagasaka, M. et al. KRAS G12C game of thrones: which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 84, 101974 (2020).
Liu, P. Y., Wang, Y. J. & Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 9, 871–879 (2019).
Riely, G. J. et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 14, 5731–5734 (2008).
Guan, J. L. et al. KRAS mutation in patients with lung cancer: a predictor for poor prognosis but not for EGFR-TKIs or chemotherapy. Ann. Surg. Oncol. 20, 1381–1388 (2013).
Thunnissen, F. B. et al. Smoking history and lung carcinoma: KRAS mutation is an early hit in lung adenocarcinoma development. Lung Cancer 75, 156–160 (2012).
Brose, M. S. et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 62, 6997–7000 (2002).
Murakami H. et al. A nationwide genomic screening project for small cell lung cancer in Japan (LC-SCRUM-Japan). ASCO 2017; Chicago, IL, USA 2017. (ASCO Annual Meeting Abstracts, Chicago, 2017).
Nassar, A. H., Adib, E. & Kwiatkowski, D. J. Distribution of KRAS G12C somatic mutations across race, sex, and cancer type. N. Engl. J. Med. 384, 185–187 (2021).
Berndt, N., Hamilton, A. D. & Sebti, S. M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791 (2011).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Canon, J. et al. The Clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumor immunity. Nature 575, 5581 (2019).
Patricelli, M. P. et al. Selective Inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 6, 316–329 (2016).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Marx M. A. et al. Structure-based drug discovery of MRTX1257, a selective, covalent KRAS G12C inhibitor with oral activity in animal models of cancer. American Association for Cancer Research, 18, 5 Supplement (2020).
Hallin, J. et al. The KRAS G12C inhibitor MRTX849 Provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020).
Janne P. A., Papadopoulos K., Ou I., Rybkin I. & Johnson M. A phase 1 clinical trial evaluating the pharmacokinetics (PK), safety, and clinical activity of MRTX849, a mutant-selective small molecule KRAS G12C inhibitor, in advanced solid tumors. AACR-NCI-EORTC (2019).
Mai, T. T. & Lito, R. A treatment strategy for KRAS-driven tumors. Nat. Med. 24, 902–904 (2018).
Cai, D., Choi, P. S., Gelbard, M. & Meyerson, M. Identification and characterization of oncogenic SOS1 Mutations in lung adenocarcinoma. Mol. Cancer Res. 17, 1002–1012 (2019).
Hillig, R. C. et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad. Sci. USA 116, 2551–2560 (2019).
Ryan, M. B. et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS G12C inhibition. Clin. Cancer Res. 26, 1633–1643 (2020).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using cBioPortal. Sci. Signal. 6, p11 (2013).
Amgen. Amgen Announces First Clinical Data Evaluating Novel Investigational KRASG12C Inhibitor AMG 510 At ASCO 2019. News Releases. https://www.amgen.com/media/news-releases/2019/06/amgen-announces-first-clinical-data-evaluating-novel-investigational-krasg12c-inhibitor-amg-510-at-asco-2019/ (2019).
Hong, D. S. et al. KRAS G12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207–1217 (2020).
B., Li. Sotorasib for KRAS G12C. World Conference on Lung Cancer (2021).
Amgen. Study to Compare AMG 510 “Proposed INN Sotorasib” With Docetaxel in Non Small Cell Lung Cancer (NSCLC) (CodeBreak 200). ClinicalTrials.gov. NIH U.S. National Library of Medicine https://www.clinicaltrials.gov/ct2/show/NCT04303780 (2021).
Skoulidis, F. et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Garon, E. B. et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet 384, 665–673 (2014).
Skoulidis, F. Overall survival and exploratory subgroup analyses from the phase 2 CodeBreaK 100 trial evaluating sotorasib in pretreated KRAS p.G12C mutated non-small cell lung cancer. ASCO (2021).
Staff, The ASCO Post. Updated information on sotorasib dose-comparison study. The ASCO Post. https://ascopost.com/news/april-2021/updated-information-on-sotorasib-dose-comparison-study/ (2021).
Janne. P. A. KRYSTAL-1: Updated safety and efficacy data with adagrasib (MRTX849) in NSCLC with KRAS G12C Mutation From a Phase 1/2 Study. AACR-NCI-EORTC (2020).
Skoulidis, F. et al. STK11/LKB1 Mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Disco. 8, 822–835 (2018).
Li B. et al. PS01.07 Registrational phase 2 trial of sotorasib in KRAS p.G12C mutant NSCLC: first disclosure of the codebreak 100 primary analysis. J. Thorac. Oncol. https://doi.org/10.1016/j.jtho.2021.01.321 (2021).
Fakih M. et al. Trial in progress: a phase Ib study of AMG 510, a specific and irreversible KRAS G12C inhibitor, in combination with other anticancer therapies in patients with advanced solid tumors harboring KRAS p.G12C mutation (CodeBreak 101). J. Clin. Oncol. https://ascopubs.org/doi/abs/10.1200/JCO.2020.38.15_suppl.TPS3661 (2020).
Reck, M. et al. 1416TiP- CodeBreak 2020: A phase III multicenter study of sotorasib (AMG 510), a KRAS(G12C) inhibitor, versus docetaxel in patients with previously treated advanced non-small cell lung cancer (NSCLC) harboring KRAS p.G12C mutation. Ann. Oncol. 31, S754–S840 (2020).
Janne, P. A. et al. 3LBA Late breaking—KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/metastatic non-small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur. J. Cancer 138, S1–S2 (2020).
Sabari J. K. et al. KRYSTAL-2: a phase I/II trial of adagrasib (MRTX849) in combination with TNO 155 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.3_suppl.TPS146 (2021).
Spira, A. et al. P90.03 a phase 2 Trial of MRTX849 in combination with pembrolizumab in patients with advanced non-small cell lung cancer with KRAS G12C mutation. J. Thorac. Oncol. 16, S696 (2021).
Brana I. et al. 3005: Initial results from a dose finding study of TNO155, a SHP2 inhibitor, in adults with advanced solid tumors. ASCO, 39, p. Suppl 15. (2021)
Gort E. et al. A phase I, open-label, dose-escalation trial of BI 1701963 as monotherapy and in combination with trametinib in patients with KRAS mutated advanced or metastatic solid tumors. J. Clin. Oncol. https://ascopubs.org/doi/abs/10.1200/JCO.2020.38.15_suppl.TPS3651 (2020).
Johnson M. L. et al. 4LBA late breaking—KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer (CRC) and other solid tumors harboring a KRAS G12C mutation. Eur. J. Cancer 138, S2 (2020).
Peng S. B. et al. 1259- Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C inhibitor. AACR Annual Meeting 2021 Novel Antitumor Agents Abstract (2021).
S., Malek. Perspective on novel approaches to drugging RAS pathway. AACR-NCI-EORTC (2020).
Inc., InventisBio. Study to evaluate D-1553 in Subjects With Solid Tumors. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04585035 (2020).
Novartis Pharmaceuticals. Study of JDQ443 in patients with advanced solid tumors harboring the KRAS G12C mutation. ClinicalTrials.gov. NIH U.S. National Library of Medicine, https://clinicaltrials.gov/ct2/show/NCT04699188 (2021).
Nichols R. J. et al. A Next Generation Tri-Complex KRAS G12C (ON) Inhibitor Directly Targets the Active, GTP-Bound State of Mutant RAS and May Overcome Resistance to KRAS G12C (OFF) Inhibition. Revolution Medicines (2021).
Revolution Medicines. Translating frontier oncology targets to outsmart cancer: corporate review Q2 2021. Revolution Medicines, (2021).
Peng S. B. et al. Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C Inhibitor. Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C Inhibitor. AACR Annual Meeting (2021).
Stinchcombe, T. E. & Johnson, G. L. MEK inhibition in non-small cell lung cancer. Lung Cancer 86, 121–125 (2014).
Li, S. et al. Assessing therapeutic efficacy of MEK inhibition in a KRAS G12C-driven mouse model of lung cancer. Clin. Cancer Res. 24, 4854–4864 (2018).
Moore, C. et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 11, 1–16 (2019).
Nichols, R. J. et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 20, 1064–1073 (2018).
Ou, S. I. et al. A12 The SHP2 inhibitor RMC-4630 in patients with KRAS-mutant non-small cell lung cancer: preliminary evaluation of a first-in-man phase 1 clinical trial. J. Thorac. Oncol. 15, S15–S16 (2020).
Therapeutics, Relay. RLY-1971 in subjects with advanced or metastatic solid tumors. ClinicalTrials.gov. NIH U.S. National Library of Medicine, https://clinicaltrials.gov/ct2/show/NCT04252339 (2020).
Jacobio Pharmaceuticals Group Co., LTD. Jacobio Pipeline. http://en.jacobiopharma.com/intro/17.html (2021).
Gerlach, D. et al. Abstract 1091: BI-3406 and BI 1701963: Potent and selective SOS1::KRAS inhibitors induce regressions in combination with MEK inhibitors or irinotecan. Cancer Res. 80, (2020). (16 Suppl):Abstract 1091.
Amgen, A. M. G. 510 (pINN) Sotorasib activity in subjects with advanced solid tumors with KRAS p.G12C mutation (CodeBreak 101). ClinicalTrials.gov. NIH U.S. National Library of Medicine, https://clinicaltrials.gov/ct2/show/NCT04185883 (2020).
Goldman, J. W. et al. A randomized phase III study of abemaciclib versus erlotinib in patients with stage IV non-small cell lung cancer with a detectable KRAS mutation who failed prior platinum-based therapy: JUNIPER. Front. Oncol. 10, 1–12 (2020).
Merck. Data from Exploratory Analysis Show Merck’s KEYTRUDA (pembrolizumab) Improved Overall Survival as Monotherapy for the First-Line Treatment of Metastatic Non-Small Cell Lung Cancer Regardless of KRAS Mutational Status. News Releases. https://www.merck.com/news/data-from-exploratory-analysis-show-mercks-keytruda-pembrolizumab-improved-overall-survival-as-monotherapy-for-the-first-line-treatment-of-metastatic-non-small-cell-lung-cancer-regard/ (2019).
Awad M., et al. LB002- Mechanisms of acquired resistance to KRAS G12C inhibition in cancer. AACR (2021).
Marcoux, N. et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J. Clin. Oncol. 37, 278–285 (2018).
Gandhi, L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018).
Jain, R. K. & Chen, H. Spotlight on brigatinib and its potential in the treatment of patients with metastatic ALK-positive non-small cell lung cancer who are resistant or intolerant to crizotinib. Lung Cancer 8, 169–177 (2017).
Douillard, J. Y. et al. Gefinitib treatment in EGFR mutated caucasian NSCLC. J. Thorac. Oncol. 9, 1345–1353 (2014).
Arcila, M. E. et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 17, 1169–1180 (2011).
Newman, A. M. et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554 (2014).
Mok, T. et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes of NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin. Cancer Res. 21, 3196–3203 (2015).
Chae, Y. K. et al. Concordance between genomic alterations assessed by next-generation sequencing in tumor tissue or circulating cell-free DNA. Oncotarget 7, 65364–65373 (2016).
Chaudhuri, A. A. et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Disco. 7, 1394–1403 (2017).
Globe Newswire. Guardant Health Announces Collaboration With Amgen to Develop a Global Liquid Biopsy Companion Diagnostic for AMG 510 KRAS G12C Inhibitor. Press Releases. Guardant Health, Inc., 2020. https://investors.guardanthealth.com/press-releases/press-releases/2020/Guardant-Health-Announces-Collaboration-With-Amgen-to-Develop-a-Global-Liquid-Biopsy-Companion-Diagnostic-for-AMG-510-KRAS-G12C-Inhibitor/default.aspx (2021).
Author information
Authors and Affiliations
Contributions
Conception and Design: G.P., F.K., K.L., and H.H. Writing, Data Collection and Figure Generation: G.P., F.K., K.L., H.H. Administrative support: H.H. and B.W.
Corresponding author
Ethics declarations
Competing interests
Hatim Husain has received research funding from Roche Sequencing Solutions, Bristol Myers Squibb, Pfizer, and Eli Lilly, honorarium from AstraZeneca, Blueprint Medicine, and Janssen, and consulting or advisor roles with Foundation Medicine, Neogenomics, Merck, AstraZeneca, Blueprints Medicine, Mirati Therapeutics, Turning Point Therapeutics, and Takeda. All other authors have no relevant disclosures to report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palma, G., Khurshid, F., Lu, K. et al. Selective KRAS G12C inhibitors in non-small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. npj Precis. Onc. 5, 98 (2021). https://doi.org/10.1038/s41698-021-00237-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00237-5
This article is cited by
-
The role of CRAF in cancer progression: from molecular mechanisms to precision therapies
Nature Reviews Cancer (2024)
-
The current landscape of using direct inhibitors to target KRASG12C-mutated NSCLC
Experimental Hematology & Oncology (2023)
-
Critical requirement of SOS1 for tumor development and microenvironment modulation in KRASG12D-driven lung adenocarcinoma
Nature Communications (2023)
-
Resistance to KRAS G12C Inhibition in Non-small Cell Lung Cancer
Current Oncology Reports (2023)
-
In vivo genome-wide CRISPR screening identifies ZNF24 as a negative NF-κB modulator in lung cancer
Cell & Bioscience (2022)
