
Abstract
Evidence supporting specific therapies for late-life treatment-resistant depression (LL-TRD) is necessary. This study used Bayesian adaptive randomization to determine the optimal dose for the probability of treatment response (≥50% improvement from baseline on the Montgomery-Åsberg Depression Rating Scale) 7 days after a 40 min intravenous (IV) infusion of ketamine 0.1 mg/kg (KET 0.1), 0.25 mg/kg (KET 0.25), or 0.5 mg/kg (KET 0.5), compared to midazolam 0.03 mg/kg (MID) as an active placebo. The goal of this study was to identify the best dose to carry forward into a larger clinical trial. Response durability at day 28, safety and tolerability, and effects on cortical excitation/inhibition (E/I) ratio using resting electroencephalography gamma and alpha power, were also determined. Thirty-three medication-free US military veterans (mean age 62; range: 55–72; 10 female) with LL-TRD were randomized double-blind. The trial was terminated when dose superiority was established. All interventions were safe and well-tolerated. Pre-specified decision rules terminated KET 0.1 (N = 4) and KET 0.25 (N = 5) for inferiority. Posterior probability was 0.89 that day-seven treatment response was superior for KET 0.5 (N = 11; response rate = 70%) compared to MID (N = 13; response rate = 46%). Persistent treatment response at day 28 was superior for KET 0.5 (response rate = 82%) compared to MID (response rate = 37%). KET 0.5 had high posterior probability of increased frontal gamma power (posterior probability = 0.99) and decreased posterior alpha power (0.89) during infusion, suggesting an acute increase in E/I ratio. These results suggest that 0.5 mg/kg is an effective initial IV ketamine dose in LL-TRD, although further studies in individuals older than 75 are required.
Similar content being viewed by others
Introduction
Monoaminergic drugs are considered first-line treatment for major depressive disorder (MDD) but can take eight weeks or longer to achieve a response (defined as ≥50% symptom improvement) [1]. Symptom improvement can be even more delayed and weaker in individuals over the age of 55 [2,3,4], a period considered “later life” [5]. There is a clear unmet need to develop and test novel psychotropics for the rapidly growing population with late-life treatment-resistant depression (LL-TRD) [6].
A single 40 min intravenous (IV) infusion of 0.5 mg/kg of the non-competitive N-methyl-D-aspartate receptor (NMDAR) antagonist ketamine (KET) can achieve antidepressant effects within 24 h in adult TRD [7]. Clinical improvement appears to strengthen with increasing doses but shows a pattern of diminishing returns above 0.5 mg/kg [7]. Clinical improvement is achieved by an initial temporary block of NMDARs located on fast firing, inhibitory GABAergic parvalbumin (PV+) interneurons with subsequent disinhibition of glutamate release that enhances the balance between cortical excitation relative to inhibition (E/I) in a prefrontal neural circuitry [8,9,10,11,12]. This change in balance between cortical excitation and inhibition can be measured with electroencephalography (EEG) as an increase in gamma oscillations [13, 14] or a decrease in alpha oscillations [15]. A successive surge of glutamate activates post-synaptic low-affinity α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs). AMPAR activation promotes intracellular brain-derived neurotrophic factor (BDNF) upregulation and a subsequent growth of dendritic spines and synaptic connectivity, extending beyond the elimination half-life of KET [8,9,10,11], potentially resulting in sustained effects on E/I balance.
Four studies, with mixed results, have examined clinical effects of KET in individuals with LL-TRD [16,17,18,19], suggesting that KET response may differ between age groups for patients with LL-TRD. Little is known about optimal, safe doses for LL-TRD, and NMDAR receptor engagement of KET in older patients. Dose finding for LL-TRD is important because of a reported decline in cortical NMDAR binding density and of channel binding sites [20], and because of possible changes in elderly populations in pharmacokinetics and pharmacodynamics [21] that could necessitate lower dosages to balance efficacy and side effects [22]. The goal of this study was to identify the best dose to carry forward into a larger clinical trial.
This study used Bayesian adaptive randomization to determine the optimal dose of KET for LL-TRD, using change in Montgomery-Åsberg Depression Rating Scale (MADRS) score at seven days post-infusion as the primary endpoint. Outcomes of a Bayesian approach provide statements that the governing value for some observed process falls within some range of values. This allows statements about the probability that treatment shows benefit of some magnitude. This data is used as the basis by the Bayesian Adaptive algorithm to change allocation of subjects or to close conditions with low probability of efficacy of a certain magnitude per pre-defined stopping rules [23]. Bayesian adaptive randomization begins with the specification of a prior distribution that formalizes the available information regarding the anticipated effect and its associated uncertainty. The prior is updated with newly observed data that results in a posterior distribution that captures all available information regarding the estimated effect size and associated uncertainty. This posterior distribution forms a new prior that is continually updated with accrual of new data. Investigators can use this process of continual updating to adapt the trial according to pre-specified rules. These include systematically altering the randomization ratios to allocate participants to more promising conditions, stopping randomization to arms meeting pre-specified futility criteria, or stopping the trial for superiority if salient pre-specified criteria are met [24].
In this double-blind study, we randomized participants with LL-TRD into one of four conditions, comparing effects of a single IV infusion of KET at 0.5, 0.25, or 0.1 mg/kg against a psychoactive control (0.03 mg/kg midazolam [MID]) on MADRS at 7 days post-infusion (primary endpoint). A 7-day primary endpoint was chosen to reflect a clinically meaningful and enduring effect.
We hypothesized that KET 0.5 mg/kg would outperform the alternative conditions at the primary endpoint. Secondary outcomes were durability of the antidepressant response at 28 days post-infusion, and safety and tolerability. Exploratory outcomes were biological and physiological markers of neural plasticity previously associated with cellular and regulatory mechanisms of KET: plasma BDNF as a proxy for cellular plasticity, and EEG gamma and alpha oscillations as biomarkers of E/I balance.
Materials and methods
The study was funded by the US Department of Veterans Affairs, monitored by a Data and Safety Monitoring Board, and registered at ClinicalTrials.gov (NCT02556606). Study procedures were approved by the Baylor College of Medicine IRB and the Michael E. DeBakey VA Medical Center Research & Development Committee. Materials and methods were described in detail in a prior publication [25] and are briefly presented here. All subjects provided written informed consent before any study-related activities were conducted. All procedures were carried out at the Michael E. DeBakey VA Medical Center in Houston, TX.
Participant selection
Participants were US military veterans at least 55 years old with DSM-5 recurrent or chronic MDD (APA, 2013); resistance to at least two adequate trials of FDA-approved antidepressants determined by the MGH Antidepressant Treatment Response Questionnaire [26]; moderate-to-severe depressive symptoms at screening and randomization (MADRS ≥ 27; Quick Inventory of Depressive Symptomatology—Self Report [QIDS-SR] ≥ 14 [27]; and Clinical Global Impression—Severity scale [CGI-S] ≥ 4 [28]). Participants were free of psychotropic medications, including antidepressants, for at least one week prior to study drug infusion. Exclusion criteria included history of psychotic disorder, bipolar disorder, substance or alcohol use disorder in the past 3 months, use of NMDAR or AMPAR medications, and any unstable medical or neurological illness.
Study design
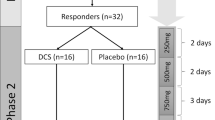
This dose-finding study used a Bayesian adaptive randomized, double-blind, placebo-controlled design. Allocation was generated adaptively using a Bayesian “bandit” paradigm by the study statistician (CEG) who was blinded to the condition. Outcomes of the most recent participant(s) were communicated by blinded study staff to the study statistician who applied pre-defined decision rules followed by re-estimation of allocation which was communicated to the pharmacy. The protocol design could stop the study for superiority, change randomization ratios, or stop the study for futility based on the probability of day-seven treatment response. Participants were initially randomized into one of two arms. One of every four participants was allocated to Arm 1, a 40-min IV infusion of 0.03 mg/kg MID. The remaining participants were allocated to Arm 2. Participants in Arm 2 were allocated 1:1:1:1 to a 40-min IV infusion of MID 0.03 mg/kg, KET 0.1 mg/kg, KET 0.25 mg/kg, or KET 0.5 mg/kg (see Supplementary Figure 1, CONSORT). Arm 2 became subject to adaptive randomization after allocation of the initial 20 participants. A condition in arm 2 was terminated if that condition demonstrated a posterior probability <0.025 that the response rate was better than the best performing condition. Arm 1 remained open for allocation to ensure a placebo group sufficiently large for comparison with the best dose of KET. For analyses, MID from Arm 1 and Arm 2 were combined. The decision to stop the study for superiority was based on the best performing condition having a posterior probability of >0.975 that it was outperforming the next best condition. The decision rules for the adaptive randomization are described further in Supplementary information.
Study procedures
Detailed descriptions of procedures are in the Supplementary information and in [25]. The Research Pharmacist prepared study drugs on the morning of infusion. All study staff and patients were blind to the condition. Study procedures on the day of infusion were in a hospital room dedicated to research. Following an overnight fast, on the morning of infusion, clinical ratings (MADRS; QIDS-SR; CGI-S), adverse events scales (Columbia-Suicide Severity Rating Scale [C-SSRS] [29], Clinician Administered Dissociation Symptom Scale [CADSS] [30]; four-item Brief Psychiatric Rating Scale [BPRS+] [31]; Patient Reported Inventory of Side Effects [PRISE] [32]), 64-channel EEG (resting eyes open and eyes closed 2 min each reported here; auditory mismatch negativity paradigm with duration deviants reported in [33]), and a blood draw for BDNF were administered before the infusion. CADSS, BPRS+ and PRISE were repeated at 40 min (end of infusion), 120 min, and 240 min after the start of infusion. EEG was repeated at 30 (resting EEG only), 60, 120, and 240 min relative to the start of infusion. The blood draw was repeated at 120, 240 min, and 8 h after the start of infusion. Blood pressure and pulse were assessed every 15 min from the start of infusion until 240 min after the start of infusion. All measures (except EEG) were repeated at 24 h (except blood collection), 48 h, 72 h, and 7 days after infusion. EEG was repeated at 24 h and 7 days after infusion. The C-SSRS was administered at all visits. Depression scales were repeated at days 14, 21, and day 28 for day-seven responders. Study staff not present at the infusion and thus blinded for possible side effects performed study procedures before and after the day of infusion.
Statistical analysis and sample size
Statistical analyses used R (v. 4.0.4) [34]. The primary endpoint was day-seven treatment response. Since the use of a binary decision-rule implies a binomial process, we started with a ~Beta(1,1) (a flat line from zero through one) for prior distributions for all conditions in arm 2 (the adaptive randomization arm). We used a Beta-Binomial model for adaptation and decision-making. Analyses for secondary and exploratory outcomes used Bayesian adaptations of generalized linear models with multilevel components for correlated data. Priors were ~Normal(µ = 0, σ = 1000) within the link function for coefficients and ~Folded T-Distribution(df = 3, µ = 0, σ = 10) for random effects and residual terms within the link function. The convergence of Bayesian analyses on the posterior distributions via Monte-Carlo Markov chain (MCMC) was assessed with Gelman-Rubin Diagnostics and Effective Sample Size estimates. Posterior distributions are described with point estimates (50th percentile) and 95% credible interval (2.5th and 97.5th percentiles).
Based on the estimates, priors, and decision rules, over K = 10,000 Monte Carlo simulations showed an average n = 5, 5, 8, and 24 participants were expected to be allocated to arm 2 MID, KET 0.1, KET 0.25, and KET 0.5 respectively to reach conclusions of superiority. For valid comparison between KET and MID, arm 1 MID remained open and we expected to allocate 18 subjects until declaration of superiority of KET 0.5. Using the prior distributions and assumed probabilities of being a day-seven responder, K = 10,000 Monte Carlo simulations indicated that the decision rules would identify the best condition (i.e., KET 0.5) 95% percent of the time; this estimate corresponds to conventional power and exceeds the widely used value of 80%.
For our primary endpoint (day-seven MADRS) we compared the change in MADRS score relative to baseline at day seven between KET 0.5 and MID. For EEG oscillatory power, we performed separate analyses to compare drug effects on frontal gamma power and posterior alpha power, respectively. For EEG we used three individual models: (1) the day 1 model investigated the effects of time (pre-infusion to 4 h post infusion) and condition (KET 0.5 vs MID) on EEG power; (2) the 24-hour model compared EEG power between conditions (KET 0.5 vs MID) at 24 h post-infusion corrected for baseline power; (3) the durability model evaluated the changes in EEG power between conditions (KET 0.5 vs MID) relative to time from the 24 h time point to day seven (24 h to 7 days, correcting for baseline power). For BDNF we used two individual models: (1) the day 1 model investigating the effect of time (pre-infusion to 4 h post infusion) and condition corrected for baseline BDNF; (2) the durability model evaluated differences between conditions at day seven corrected for baseline BDNF.
Results
Bayesian adaptive randomization implemented after enrolling the first 20 participants to Arm 2 terminated allocation to MID, KET 0.1 and KET 0.25 in that arm. Stopping rules terminated trial enrollment after completing 33 participants. Supplementary Figure 1 presents the allocation of participants. Table 1 presents demographic and clinical characteristics.
Primary endpoint
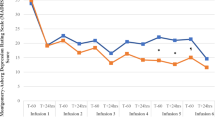
Figure 1 presents MADRS total scores as a function of condition and time (panel A), proportions of participants with a treatment response defined as an improvement by at least 50% from baseline (panel B), and posterior distributions of the probability of a day-seven treatment response (panel C). The information depicted in panel 1C represents both our best estimate of the effects and the associated uncertainty which results, from among other things, small cell sizes. The comparison of the posterior distributions (subtracting one distribution from the other) yields the probability that one distribution is higher or lower than the other.

MADRS total scores (± standard error) as a function of condition and time from pre-infusion baseline to day-seven post-infusion (A); proportions of patients with a treatment response (≥ 50% improvement in MADRS from baseline) as a function of condition and time (B); Bayesian posterior distributions of probabilities of a day-seven treatment response as a function of condition (C).
Sixteen of 33 randomized participants (8 for KET 0.5; 2 for KET 0.25; 5 for MID) achieved a day-seven treatment response; all day-seven responders but one (MID) achieved day-seven remission (see Supplementary information). KET 0.5 showed an absolute probability of achieving a day-seven treatment response of 0.70 (95%-CrI = 0.43–0.90). The evidence was strong that the probability of achieving a day-seven treatment response was higher for KET 0.5 than for MID (0.46, 95%-CrI = 0.23–0.71) (posterior probability [KET 0.5 > MID] = 0.89). Inspection of posterior probabilities for effects of increasing magnitude suggested moderately high evidence (0.76) that the probability of achieving a day- seven treatment response was at least 10% higher for KET 0.5 than for MID, but less so that the two conditions differed by at least 20% (posterior probability = 0.57). KET 0.25 showed minimal evidence of differing from MID (Posterior Probability [KET 0.25 > MID] = 0.42), whereas probability was high that KET 0.1 had a lower probability of achieving a day-seven treatment response than MID (posterior probability [KET 0.1 > MID] = 0.07). Treatment-related changes on QIDS-SR and CGI-S are displayed in Supplementary Fig. 2.
Durability
KET 0.5 had superior response durability. Of the 16 patients with a day-seven treatment response, 7 of 8 for KET 0.5, 1 of 2 patients for KET 0.25, and 2 of 6 patients for MID continued meeting response criteria at day 28. For remission, sustained remission was found in 6 of the 8 day-seven remitters for KET 0.5, 1 of the 2 day-seven remitters for KET 0.25, and 2 of the 5 day-seven remitters for MID. KET 0.5 responders at day-seven had high absolute probability of being day 28 responders (0.82, 95%-CrI = 0.52–0.97). KET 0.25 and MID day-seven responders had moderate (0.50, 95%-CrI = 0.09–0.90) and low (0.37, 95%-CrI = 0.10–0.71) absolute probabilities of being day 28 responders respectively. The broad credible intervals for those two conditions showed the high degree of uncertainty about the distribution and should be interpreted with caution.
Safety and tolerability
Pre-existing medical conditions are provided in Supplementary Table 1. KET 0.5 had a transient and dose-dependent increase in dissociative symptoms on the CADSS, as well as systolic and diastolic blood pressure, and pulse around end of infusion (Supplementary Fig. 3); there was no notable change in BPRS+. The probabilities of side effects on body systems assessed with the PRISE were moderate to low across interventions (Supplementary Table 2). One participant made a suicide attempt four days after MID infusion; this was determined to be unrelated to the intervention as this participant scored zero on the C-SSRS one day before the attempt.
Resting state EEG
The complete analytical report is summarized in Supplementary Table 3. EEG analyses focused on the KET 0.5 (N = 8) and MID (N = 9) arms, given the small sample sizes in the two lowest KET dose arms. Figure 2 presents frontal gamma power (panel A), posterior alpha power (panel B), and posterior alpha peak frequency (panel C) as functions of condition and time.

EEG (±standard error) as a function of condition and time for ketamine 0.5 mg/kg (KET 0.5) and midazolam (MID). Displayed are frontal gamma power (A), posterior alpha power (B), and posterior alpha peak frequency (C).
Frontal gamma power
Day 1 model: analysis showed strong evidence for a time-by-condition interaction for gamma power (posterior probability [time × condition interaction > 0] = 0.92). KET 0.5 showed an initial increase in gamma power at end of infusion followed by a decrease continuing until the 240-min measurement (posterior probability [KET 0.5 < 0] = 0.99; see Supplementary Table 3) (Fig. 2A). MID had high probability of an increase over time (posterior probability [MID > 0] = 0.91).
24-h model: at 24 h, frontal gamma power appeared comparable for KET 0.5 and MID (posterior probability [KET 0.5 > MID] = 0.370). Durability model: posterior distributions of the time-by-condition interaction for model 3 could not be estimated by the model. However, there was moderate certainty of an increase in gamma power between 24 hours and day-seven for KET 0.5 and MID.
Posterior alpha power
Day 1 model: there was little evidence of a time-by-condition interaction for alpha power (posterior probability [time × condition interaction > 0] = 0.571). There was strong evidence of an initial decrease followed by an increase in alpha power for KET 0.5 and MID (Fig. 2B).
24-h model: KET 0.5 had high probability of greater alpha power than MID (posterior probability [KET 0.5 > MID] = 0.968). Durability model: evidence was weak for a time-by-condition interaction for change in alpha power between 24 hours and day-seven (posterior probability [time × condition interaction > 0] = 0.082).
Posterior alpha peak frequency
Day 1 model: analysis showed moderate evidence for a time-by-condition interaction for alpha peak frequency (posterior probability [time × condition interaction < 0] = 0.763). Posterior alpha peak frequency showed an initial increase followed by a decrease for KET 0.5 (Fig. 2C) although the evidence for this increase was weak (posterior probability [KET 0.5 < 0] = 0.558). Evidence for an initial decrease followed by an increase in alpha peak frequency for MID was moderately strong (posterior probability [MID > 0] = 0.778).
24-h model: the evidence was inconclusive about whether alpha peak frequencies at 24 hours differed between KET 0.5 and MID (posterior probability [KET 0.5 > MID] = 0.551). Durability model: posterior distributions for alpha peak frequency slopes between 24 h and day-seven could not be estimated by the model.
BDNF
The complete analytical report is summarized in Supplementary Table 4. Supplementary Fig. 4 displays plasma BDNF concentrations as a function of condition and time. Day 1 model: The evidence for an interaction between time and condition on day one was weak (posterior probability = 0.57). Testing simple effects of time per condition revealed a high probability of increased BDNF as a function of time for KET 0.5 (posterior probability [KET 0.5 > 1] = 0.94). Probability was weak for a decrease as a function of time for MID (posterior probability [MID < 1] = 0.60) but moderate for KET 0.25 (posterior probability [KET 0.25 < 1] = 0.77). The posterior distribution could not be estimated by the model for KET 0.1. Durability model: At day-seven, the evidence was weak that geometric mean of plasma BDNF was lower for KET 0.5 than MID (posterior probability [KET 0.5 < MID] = 0.63). Conversely, there was strong evidence that the geometric mean was higher for KET 0.25 (posterior probability [KET 0.25 > MID] = 0.95) and KET 0.1 (posterior probability [KET 0.1 > MID] = 0.85) than MID.
Exploratory correlational analysis
In a recent study baseline gamma power was shown to moderate clinical response to KET at 230 min after the start of infusion in TRD patients [35]. The authors posit that this could reflect a greater susceptibility to improvements in cortical homeostasis brought on by KET, with patients that experience the greatest baseline deficits having the clearest gains from treatment. We conducted an exploratory analysis to determine whether MADRS outcomes for KET 0.5 and MID were related to gamma power reactivity (the difference between peak gamma during infusion and baseline). Greater gamma reactivity was associated with a stronger reduction in MADRS at 7 days post infusion for KET (τ = −0.5, p = 0.08), but no relationship was observed for midazolam (τ = 0.05, p = 0.8) (Fig. 3) (see Supplementary Table 5).

Ketamine demonstrated a moderate relationship between gamma reactivity at 30 min and change in MADRS score at 7 days post infusion. This relationship was less prevalent at the earlier time points and was non-existent for patients in the midazolam condition. More positive MADRS change scores (x-axes) indicates smaller decreases/an increase in MADRS from baseline. Correlation results are summarized in Supplementary Table 5.
Discussion
In this study, we addressed dose optimization for KET treatment of LL-TRD. Using Bayesian adaptive randomization we compared the treatment response at day seven after single infusions of IV KET at three doses (0.5, 0.25, 0.1 mg/kg) relative to midazolam (0.03 mg/kg) in a sample with a mean age of 62 (range: 55–72 years old). KET 0.5 proved superior to the other conditions. Bayesian adaptive randomization terminated allocation of participants to KET 0.25 and 0.1. There was a 70% chance that participants administered KET 0.5 achieved a treatment response at day-seven post-infusion, compared to a 46% chance for MID. Evidence was strong that the probability to achieve a day-seven treatment response was at least 10% higher for KET 0.5 than MID. Further, evidence was strong that responders to KET 0.5 at day-seven remained responders at day 28 follow-up. These outcomes are broadly consistent with a dose-response trial in younger adults with TRD (mean age of 44.38) that showed antidepressant superiority for KET 0.5 relative to lower doses (0.2 and 0.1 mg/kg) and to MID at 24 hours after a single infusion [36]. Adverse events for KET 0.5 had low posterior probabilities, indicating that the safety and tolerability of KET 0.5 in our sample were generally acceptable. These outcomes require further confirmation in an adequately powered trial.
Although treatment response was the primary endpoint, a 50% improvement is often inadequate for functional recovery. We showed that at seven days post-infusion all eight KET 0.5 responders also met criteria for remission. Of the eight remitters, six were still in remission at 28 days post-infusion. These findings indicate that an initial remission to KET at 0.5 mg/kg is a positive indicator of possible sustained remission at later measurements.
Increased power in the gamma frequency band of the EEG is a translational biomarker of NMDAR block on PV+ interneurons [37] and associated in animals with a block of NMDAR subunits NR1 [38] and NR2A [39]. There was a sharp increase in gamma power over the course of KET infusion which then gradually reset over a period of four hours, indicating enhanced cortical excitability relative to inhibition after blockade of NMDAR, aligning with outcomes of other NMDAR antagonists [14, 40]. At 24-h follow-up, gamma power was comparable to baseline in both conditions, with weak evidence of a shared increase at day-seven. This suggests acutely enhanced cortical excitation related to blocking NR1/NR2A, without sustained KET-related changes. However, the lower strength of evidence makes it impossible to rule out changes in gamma at day-seven being related to biological or measurement fluctuations. In earlier work baseline gamma power moderated clinical response to KET at 230 minutes [35]. Relationships between gamma reactivity at 30 minutes from baseline and clinical response showed a negative relationship between increased reactivity and decreased depression severity at day-seven post-infusion for KET 0.5 but not MID. This suggests that stronger engagement of KET with the NR1/NR2A subunits could initiate biological processes leading to rapid antidepressant effects persisting for at least one week. While these findings should be interpreted cautiously due to the small sample size and exploratory nature, this might indicate preliminary support for the utility of EEG gamma power as a predictive biomarker for KET clinical response. Future larger studies using an appropriate longitudinal design are required for confirmation.
Resting state alpha oscillations provide information about the ability of an upstream system to exert control over the gain measured in downstream local neuronal clusters where a reduction in power reflects enhanced excitability of the local cortex [41]. We found strong evidence that KET 0.5 and MID suppressed posterior resting state alpha power at the end of infusion consistent with prior studies [15], possibly indicating an instantaneous increase in neural excitability [42]. Decrease in alpha power after KET has been associated in animals with blockade of NMDAR subunit NR2B [43], possibly by increasing cholinergic activity [44]. It is likely that MID could behave similarly, but through non-NMDAR mechanisms. Resting alpha power was lower for MID than KET 0.5 at 24 hours post-infusion, possibly reflecting sustained cholinergic inhibition in MID, without sustained effect of KET 0.5. Effects for alpha power from 24 hours to day-seven could not be estimated by the statistical model. Evidence was moderate that alpha had a lower peak frequency for MID, but was inconclusive on whether alpha peak frequency changed with KET 0.5. This suggests acutely enhanced cortical excitation related to blockade of NR2B without sustained KET-related changes.
We investigated plasma BDNF as a peripheral biological marker of KET-associated neuroplasticity [8,9,10,11]. The evidence was low that conditions differed in BDNF response which suggest that further study is warranted to accurately explore possible dose-response effect in LL-TRD (see Supplementary material).
Counter to expectations, approximately 50% of patients randomized to MID met treatment response criteria on day seven, though MID had a low probability of a sustained antidepressant effect at day 28. Although this may be a placebo effect, MID could also have antidepressant properties, even if durability would be shorter than KET. MID is a positive allosteric modulator of GABAA receptors located on glutamate receptors, inhibiting glutamate release; KET creates a glutamate surge. Two possible explanations are (1) antidepressant effects of KET might involve simultaneous block of NMDARs and activation of GABAA receptors [45], implying that responders to MID and KET represent a single biotype; (2) MID and KET may have opposite effects on glutamate release and consequently on excitability of pyramidal neurons, implying that responders to MID and KET represent two different biotypes [45]. This latter hypothesis could be partially consistent with diminished GABAA receptor-associated cortical inhibition, measured with paired-pulse transcranial magnetic stimulation as diminished short intracortical inhibition for young and elderly people with depression, compared to young, but not elderly, healthy controls [46].
A strength of the current study was the use of Bayesian methods to determine superiority across doses by terminating allocation of participants to treatment arms with low efficacy, potentially resulting in more efficient and less costly trials if one intervention arm is clearly superior. This approach may be more ethical than traditional designs because it limits exposure of patients to interventions with low efficacy. Another strength was the enrollment of psychotropic medication-free TRD patients, permitting interpretation of treatment response and neurophysiological measures uncontaminated by concurrent psychotropic medication use.
Limitations of this study include (1) patients were U.S. military veterans and predominantly male, which may limit generalization of outcomes to non-veterans and to women; (2) lack of enrollment of participants from the oldest age cohort (>75 years); in a recent phase 3 trial of intranasal esketamine in late-life TRD [19], patients 75 and older did not show benefit relative to placebo; (3) we did not examine relationships between changes in depression severity and blood levels of KET or its metabolite norketamine, which correlated with improvement in depression in adult TRD [47]; (4) the outcomes of EEG and BDNF should be interpreted with caution due to small sample sizes and the use of midazolam instead of inert saline placebo; (5) the Bayesian adaptive randomization scheme denied the two poor performing KET arms (0.1, 0.25) adequate sample sizes to conduct meaningful BDNF and EEG analyses; (6) the relationship between change in MADRS with KET-induced gamma band reactivity requires further examination in larger samples to determine how well reactivity could be a biomarker of the magnitude of improvement in depression; and (7) aspects of illness course may have influenced response across treatment conditions, and further study could examine those effects. This highlights a design limitation where the clinical trial structure is not conducive to simultaneous collection of data in adequate quantity for detailed understanding of biomarkers. This distinction between experimental styles should be carefully considered by future studies to ensure sufficient data collection for biomarkers in early-stage drug discovery programs. On the other hand, this adaptive strategy provides earlier identification of effective compounds for subsequent neurobiological studies.
In conclusion, a single infusion of KET 0.5 mg/kg was superior to lower doses and midazolam in achieving a treatment response at day-seven and in maintaining a treatment response up to day 28. We were unable to detect meaningful effects at the same time points for EEG gamma and alpha power suggesting that those physiological measures are not biomarkers for sustained biological changes directly associated with treatment response. Future studies should investigate optimal dosing strategies in conjunction with relevant physiological biomarkers to determine the sensitivity of biological marker detection for both acute and sustained clinical responses. The optimal KET treatment schedule for LL-TRD awaits further study.
References
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40.
Nelson JC, Delucchi K, Schneider LS. Efficacy of second generation antidepressants in late-life depression: a meta-analysis of the evidence. Am J Geriatr Psychiatry. 2008;16:558–67.
Zanardi R, Cusin C, Rossini D, De Ronchi D, Serretti A. Comparison of response to fluvoxamine in nondemented elderly compared to younger patients affected by major depression. J Clin Psychopharmacol. 2003;23:535–9.
Tedeschini E, Levkovitz Y, Iovieno N, Ameral VE, Nelson JC, Papakostas GI. Efficacy of antidepressants for late-life depression: a meta-analysis and meta-regression of placebo-controlled randomized trials. J Clin Psychiatry. 2011;72:1660–8.
Beekman ATF, Copeland J, Prince MJ. Review of community prevalence of depression in later life. Br J Psychiatry. 1999;174:307–11.
Heo M, Murphy CF, Fontaine KR, Bruce ML, Alexopoulos GS. Population projection of US adults with lifetime experience of depressive disorder by age and sex from year 2005 to 2050. Int J Geriatr Psychiatry. 2008;23:1266–70.
Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, Bauer M, et al. Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med. 2016;46:1459–72.
Abdallah CG, Sanacora G, Duman RS, Krystal JH. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacol Ther. 2018;190:148–58.
Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, Chowdhury GMI, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology. 2018;43:2154–60.
Abdallah CG, Jackowski A, Salas R, Gupta S, Sato JR, Mao X, et al. The nucleus accumbens and ketamine treatment in major depressive disorder. Neuropsychopharmacology. 2017;42:1739–46.
Gilbert JR, Zarate CA. Electrophysiological biomarkers of antidepressant response to ketamine in treatment-resistant depression: gamma power and long-term potentiation. Pharmacol Biochem Behav. 2020;189:172856.
Fagerholm ED, Leech R, Williams S, Zarate CA, Moran RJ, Gilbert JR. Fine-tuning neural excitation/inhibition for tailored ketamine use in treatment-resistant depression. Transl Psychiatry. 2021;11:335.
Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25.
Murphy N, Ramakrishnan N, Vo-Le B, Vo-Le B, Smith MA, Iqbal T, et al. A randomized cross-over trial to define neurophysiological correlates of AV-101 N-methyl-D-aspartate receptor blockade in healthy veterans. Neuropsychopharmacology. 2021;46:820–7.
Lozano-Soldevilla D. On the physiological modulation and potential mechanisms underlying parieto-occipital alpha oscillations. Front Comput Neurosci. 2018;12:23.
Szymkowicz SM, Finnegan N, Dale RM. Failed response to repeat intravenous ketamine infusions in geriatric patients with major depressive disorder. J Clin Psychopharmacol. 2014;34:285–6.
Lipsitz O, Di Vincenzo JD, Rodrigues NB, Cha DS, Lee Y, Greenberg D, et al. Safety, tolerability, and real-world effectiveness of intravenous ketamine in older adults with treatment-resistant depression: a case series. Am J Geriatr Psychiatry. 2021:S1064748121000026.
George D, Gálvez V, Martin D, Kumar D, Leyden J, Hadzi-Pavlovic D, et al. Pilot randomized controlled trial of titrated subcutaneous ketamine in older patients with treatment-resistant depression. Am J Geriatr Psychiatry. 2017;25:1199–209.
Ochs-Ross R, Daly EJ, Zhang Y, Lane R, Lim P, Morrison RL, et al. Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment-resistant depression-TRANSFORM-3. Am J Geriatr Psychiatry. 2020;28:121–41.
Magnusson KR, Brim BL, Das SR. Selective vulnerabilities of N-methyl-D-aspartate (NMDA) receptors during brain aging. Front Aging Neurosci. 2010;2:11.
Boyce RD, Handler SM, Karp JF, Hanlon JT. Age-related changes in antidepressant pharmacokinetics and potential drug-drug interactions: a comparison of evidence-based literature and package insert information. Am J Geriatr Pharmacother. 2012;10:139–50.
Holper L. Optimal doses of antidepressants in dependence on age: combined covariate actions in Bayesian network meta-analysis. EClinicalMedicine. 2020;18:100219.
May Lee K, Lee JJ. Evaluating Bayesian adaptive randomization procedures with adaptive clip methods for multi-arm trials. Stat Methods Med Res. 2021;30:1273–87.
Berry DA. Bayesian clinical trials. Nat Rev Drug Discov. 2006;5:27–36.
O’Brien B, Green CE, Al-Jurdi R, Chang L, Lijffijt M, Iqbal S, et al. Bayesian adaptive randomization trial of intravenous ketamine for veterans with late-life, treatment-resistant depression. Contemp Clin Trials Commun. 2019;16:100432.
Chandler GM, Iosifescu DV, Pollack MH, Targum SD, Fava M. Validation of the Massachusetts General Hospital Antidepressant Treatment History Questionnaire (ATRQ). CNS Neurosci Ther. 2010;16:322–5.
Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54:573–83.
Guy W. ECDEU assessment manual for psychopharmacology. Rockville, MD: US Department of Health, Education, and Welfare Public Health Service Alcohol, Drug Abuse, and Mental Health Administration; 1976.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–77.
Bremner JD, Krystal JH, Putman FW, Southwick SM, Marmar CR, Charney DS, et al. Measurement of dissociative states with the Clinican-Administered Dissociative States Scale (CADSS). J Trauma Stress. 1998;11:125–36.
Overall JE, Gorham DR. The brief psychiatric rating scale. Psychol Rep. 1962;10:799–812.
Rush AJ, O’Neal BL. Patient Rated Inventory of Side Effects (PRISE): unpublished rating scale. Dallas: University of Texas Southwestern Medical Center; 1999.
Murphy N, Lijffijt M, Ramakrishnan N, Vo-Le B, Vo-Le B, Iqbal S, et al. Does mismatch negativity have utility for NMDA receptor drug development in depression? Braz J Psychiatry. 2021. In press. https://doi.org/10.1590/1516-4446-2020-1685.
R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2019.
Nugent AC, Ballard ED, Gould TD, Park LT, Moaddel R, Brutsche NE, et al. Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Mol Psychiatry. 2019;24:1040–52.
Fava M, Freeman MP, Flynn M, Judge H, Hoeppner BB, Cusin C, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2018;25:1592–603.
Sullivan EM, Timi P, Hong LE, O’Donnell P. Reverse translation of clinical electrophysiological biomarkers in behaving rodents under acute and chronic NMDA receptor antagonism. Neuropsychopharmacology. 2015;40:719–27.
Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–69.
Kocsis B. Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatry. 2012;71:987–95.
Ramakrishnan N, Lijffijt M, Green CE, Balderston NL, Murphy N, Grillon C, et al. Neurophysiological and clinical effects of the NMDA receptor antagonist lanicemine (BHV-5500) in PTSD: A randomized, double-blind, placebo-controlled trial. Depress Anxiety. 2021;38:1108–19.
Chaumon M, Busch NA. Prestimulus neural oscillations inhibit visual perception via modulation of response gain. J Cogn Neurosci. 2014;26:2514–29.
Iemi L, Chaumon M, Crouzet SM, Busch NA. Spontaneous neural oscillations bias perception by modulating baseline excitability. J Neurosci. 2017;37:807–19.
Banerjee P, Donello JE, Hare B, Duman RS. Rapastinel, an NMDAR positive modulator, produces distinct behavioral, sleep, and EEG profiles compared with ketamine. Behav Brain Res. 2020;391:112706.
Schumacher J, Thomas AJ, Peraza LR, Firbank M, Cromarty R, Hamilton CA, et al. EEG alpha reactivity and cholinergic system integrity in Lewy body dementia and Alzheimer’s disease. Alzheimers Res Ther. 2020;12:46.
Fogaça MV, Duman RS. Cortical GABAergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci. 2019;13:87.
Lissemore JI, Bhandari A, Mulsant BH, Lenze EJ, Reynolds CF, Karp JF, et al. Reduced GABAergic cortical inhibition in aging and depression. Neuropsychopharmacology. 2018;43:2277–84.
Farmer CA, Gilbert JR, Moaddel R, George J, Adeojo L, Lovett J, et al. Ketamine metabolites, clinical response, and gamma power in a randomized, placebo-controlled, crossover trial for treatment-resistant major depression. Neuropsychopharmacology. 2020;45:1398–404.
Acknowledgements
The opinions expressed reflect those of the authors and not necessarily those of the Department of Veterans Affairs, or the U.S. government. This work is supported by a Department of Veterans Affairs Merit Award (Grant # CX-001205-01AI), and by facilities and resources of the Michael E DeBakey VA Medical Center.
Author information
Authors and Affiliations
Contributions
The study was conceptualized by SJM, RAJ, ACS, CEG, and ML. Study procedures and data collection were performed by SI, LCC, TI, ML, SJM, ACS, LH, and DAF. Data pre-processing and analysis was performed by CG, NM, CNH, and NR. Interpretation of the data was performed by SJM, CEG, ML, NM, and CNH. The first draft of the article was written by ML, NM, and SJM. All authors contributed to the drafting and revising of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
SJM has served as a consultant to Allergan, Alkermes, Axsome Therapeutics, BioXcel Therapeutics, Clexio Biosciences, Eleusis, EMA Wellness, Engrail Therapeutics, Greenwich Biosciences, Intra-Cellular Therapies, Janssen, Levo Therapeutics, Perception Neurosciences, Neurocrine, Relmada Therapeutics, Sage Therapeutics, Seelos Therapeutics, and Signant Health. He has served as an investigator for studies funded by Janssen, Merck, NeuroRx, and Sage Therapeutics, and has received research support from Biohaven Pharmaceuticals and VistaGen Therapeutics. RKA has served on the Janssen advisory and speaker board. ML has served as principal investigator for trials funded by NeuroRx and VistaGen Therapeutics. The remaining authors have nothing to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lijffijt, M., Murphy, N., Iqbal, S. et al. Identification of an optimal dose of intravenous ketamine for late-life treatment-resistant depression: a Bayesian adaptive randomization trial. Neuropsychopharmacol. 47, 1088–1095 (2022). https://doi.org/10.1038/s41386-021-01242-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01242-9
This article is cited by
-
Treating intrusive memories after trauma in healthcare workers: a Bayesian adaptive randomised trial developing an imagery-competing task intervention
Molecular Psychiatry (2023)
-
Neural complexity EEG biomarkers of rapid and post-rapid ketamine effects in late-life treatment-resistant depression: a randomized control trial
Neuropsychopharmacology (2023)
-
International pooled patient-level meta-analysis of ketamine infusion for depression: In search of clinical moderators
Molecular Psychiatry (2022)
