
Abstract
The high brilliance of ultrashort X-ray pulses recently generated in free electron lasers will soon open the way to the investigation of non-linear processes that still remain inaccessible due to the smallness of the corresponding cross sections. One of them is stimulated Compton scattering from molecules. In this work, we investigate stimulated Compton scattering from fixed-in-space H2 molecules in the few-hundred eV photon energy range, where both dipole and non-dipole transitions are important. We show that the interference between dipole and non-dipole transitions leads to pronounced asymmetries in the electron angular distributions. These asymmetries strongly depend on molecular orientation, to the point that they can lead to electron emission in either the forward or the backward directions with respect to the propagation axis, or in both directions, or even in the orthogonal direction. This is in contrast with Compton scattering from free electrons or atomic targets.
Similar content being viewed by others
Introduction
Kinematically complete experiments at synchrotron radiation facilities have recently made it possible to observe Compton scattering of energetic photons from a helium target with unprecedented detail1, thus providing an additional tool, so far unexplored, to investigate electron dynamics in atoms and molecules. These are very challenging experiments, since, in addition to the inherent complication of detecting the various charged fragments in coincidence with good enough statistics2, Compton scattering cross-sections are extremely small: around six orders of magnitude smaller than photoionization cross-sections associated with the absorption of that same photon1,3,4,5,6,7,8,9. This is practically an unsurmountable barrier for molecules, as one must also account for the additional nuclear degrees of freedom.
Due to their high brilliance and short duration, X-ray free-electron laser (XFEL) pulses10,11,12,13,14,15,16,17 should be the ideal tool to investigate Compton scattering from molecules in the few-hundred eV to keV photon energy range. Indeed, ultrashort pulses currently generated by XFELs15,18 are at least ten orders of magnitude more intense than those generated in synchrotron radiation facilities, so that they can efficiently compensate for the smallness of the cross-sections by providing a much larger number of photons in a short time interval. Because of this, XFELs have already opened the way to the observation of non-linear (multi-photon) processes in the X-ray wavelength spectral region5,19,20,21,22,23,24, to image single-molecule structural changes by means of photoelectron diffraction25 and X-ray diffraction26, or to pump and probe electron dynamics occurring in valence and inner-valence molecular states with few-femtosecond resolution27,28,29,30,31,32,33. The emerging high repetition rate at several XFEL facilities will further facilitate the investigation of such elusive processes by using multi-particle coincidence spectroscopy.
Compton scattering of photons by electrons bound to atoms differs from standard Compton scattering, i.e., from scattering by quasi-free electrons, in that it is a three-body problem instead of a two-body one. In standard Compton scattering, momentum exchange between the incoming photon and the free-electron governs the whole process. In Compton scattering by bound electrons, an electron is ejected as a result of the photon kick, and the remaining ion recoils to satisfy momentum conservation1,7. Determining the momentum of the scattered photon thus requires the joint measurement of the electron and ion momenta.
An interesting observation in the above-mentioned He experiment1, performed with photons of keV energy, was that, as a consequence of the electron binding energy, photons are not scattered in the forward direction. The reason for this behavior is that, at keV energy, the photon-atom interaction potential is dominated by the non-dipole A2 term instead of by the usual dipole A ⋅ P term, where P is the electron momentum and A is the vector potential of the incident light.
The choice of keV photon energies to perform such experiments in He was not capricious, as the contribution of the A2 term rapidly decreases with decreasing photon energy, thus leading to a further reduction of the cross section34. However, at sub-keV photon energies, the contributions from A2 and A ⋅ P can eventually become comparable in magnitude7,35. How this could affect Compton scattering remains an open question, although, as A2 and A ⋅ P couple the initial electronic state with states of different symmetries, interferences leading to unusual electron emission should be expected. One can anticipate an even richer scenario in the case of molecular targets36, since, as is well known from multi-coincidence photoionization experiments performed at lower photon energies, electron angular distributions are strongly dependent on molecular orientation (see, e.g.37,38,39).
Motivated by the exceptional capabilities of XFELs, in this work we have studied Compton scattering from fixed-in-space H2 molecules in the photon energy range where both A2 and A ⋅ P terms are comparable in magnitude (~500 eV). As pulses generated at XFELs are rapidly reaching sub-femtosecond duration28,30,32,40 (in order to further increase their intensity), the incoming radiation is far from being monochromatic. In this scenario, the inelastic scattering of an incoming photon can be stimulated by the presence of photons with the energy of the emitted photon (stimulated Compton scattering, SCS). We note that the necessary energy to overcome the H2 ionization potential for maximum momentum transfer in standard photon (Compton) scattering from an electron at rest (photon backscattering) is ~2 keV. Here we show that SCS is quite apparent at ~500 eV, far below this threshold value, in spite of the fact that, at this energy, only the high momentum tail of the bound electron momentum distribution can be ionized. Somehow, SCS reminds one of stimulated X-ray Raman scattering (SXRS) already observed at LCLS41,42 and Eu-XFEL24, except for the fact that stimulated emission of light in SXRS is not accompanied by ionization. We show that, in SCS at ~500 eV, the combination of the A2 and A ⋅ P terms gives rise to asymmetric electron angular distributions and that, for a given photon incidence direction and a given photon-scattering angle, this asymmetry strongly depends on molecular orientation. The magnitude of these asymmetries is associated with specific photon-scattering angles. Therefore, the additional knob provided by molecular orientation in the few-hundred eV photon energy range, in combination with light-incidence and polarization directions, provides a rich scenario for electron emission beyond that found in photoionization experiments performed at low-photon energies.
Results and discussion
Theory
For the ultrashort pulse durations considered in this work (200 attoseconds), chosen to mimic those achievable at XFELs28,32,40, and in the absence of autoionizing resonances, the nuclei will not have time to move before the electron is ejected. Therefore, we will ignore the nuclear degrees of freedom and will work in the fixed-nuclei approximation. We note however that, for longer pulses, e.g., of the order of a few femtoseconds or more, the inclusion of the nuclear degrees of freedom is mandatory, as hydrogen atoms are very light and can move considerably in such a short time interval43. In the fixed-nuclei approximation, the hamiltonian of the H2 molecule can be written as
where \({{{{{{{{\mathcal{H}}}}}}}}}_{el}\) is the electronic Hamiltonian of the field-free H2 molecule. We will consider two cases: interaction with a single linearly polarized pulse (one color, A) and with two linearly polarized pulses (two colors, A = A1+A2). The first case will allow us to describe Compton scattering in which the directions of the incoming and outgoing photons are the same, i.e., no deviation of the scattered photon with respect to the incidence direction35,44. In the second case, the directions of the incoming and outgoing photons can be different if the propagation directions of the two pulses are also different. As an illustration, here we will consider the case of counter-propagating pulses, which will allow for backward Compton scattering of the incoming photon. Intermediate situations can be considered by varying the relative angle between the two propagation directions7. In either case, at energies ~500 eV, the dipole terms, A(ri, t) ⋅ Pi, can be safely treated within the dipole approximation7,45,46 and the non-dipole terms, A2(ri, t), to first order in (k1 − k2) ⋅ ri, where k1 and k2 are the momenta of the incoming and outgoing photons, respectively, ki = ωini/c, with ni the light-incidence direction7,8,35,44. Under these approximations, the Hamiltonian can be written as
where ϵ is the light polarization direction, which is assumed to be the same for both pulses, and Ak(t) = \({A}_{k}^{0}f(t)\cos ({\omega }_{k}t)\), being \({A}_{k}^{0}\) the vector potential amplitude and f(t) the pulse temporal envelope (Gaussian envelope in this work). Notice that, in this expression, we have removed the \({[{A}_{1}(t)+{A}_{2}(t)]}^{2}\) term by performing the appropriate gauge transformation and that only interaction terms up to order 1/c have been retained, which is an excellent approximation for the low momentum transfer (k1−k2) expected at these energies, irrespective of the directions of k1 and k2. Notice also that, in the single-pulse case, there is no sum over the indexes j and k. The validity of these approximations has been demonstrated by Dondera et al.8 in their study of SCS from atomic hydrogen. In that work, results obtained by including higher-order terms were compared with those obtained by only keeping the same lower-order terms as in the present work. No significant differences between the two sets of calculations were observed, even though the photon energy considered in those calculations was 50 a.u. and, therefore, non-dipole effects should be more prominent than in our case (15–20 a.u. photon energy).
We have solved the time-dependent Schrödinger equation by using this approximate Hamiltonian (2) within the spectral method. A detailed description of this method can be found elsewhere45,47,48. In brief, the time-dependent wave function is expanded on a basis of 6880 correlated two-electron states described as linear combinations of anti-symmetrized products of H\({\,\!}_{2}^{+}\) orbitals represented in a basis of radial B-spline functions and spherical harmonics43. The angular momentum expansion is truncated at ℓ = 16. The B-spline radial box extends up to 60 a.u. and includes 280 of these functions. The typical number of configurations used for continuum states is 280 and for bound states, it varies between 390 and 700. The continuum states included in the expansion satisfy the proper incoming scattering boundary conditions.
Ionization probabilities
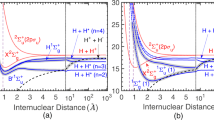
Figure 1a shows a sketch of the SCS process for the single-pulse case (central frequency 20 a.u., intensity 1018 W/cm2, and duration 200 attoseconds). Here, SCS proceeds through the absorption of a photon and the emission of a lower-energy photon in the direction of the incoming photon. This is possible because both photon energies are contained in the pulse bandwidth, which is of the order of 2 a.u. Figure 1b shows the corresponding ionization probabilities for three typical orientations of the molecule with respect to the photon incidence direction. As can be seen in the left panel of Fig. 1b, when the molecule is parallel to the polarization direction (hence, perpendicular to the light-incidence direction), the ionization probability is dominated by the non-dipole A2 terms, but this is no longer true when the molecule is perpendicular to the polarization direction (see middle and right panels of Fig. 1b), irrespective of the light-incidence direction (parallel or perpendicular to the molecular axis). In this case, dipole A ⋅ P and non-dipole A2 contributions are comparable in magnitude for electron energies close to the threshold, but the dipole contribution becomes increasingly dominant as the electron energy increases.

a Sketch of the stimulated Compton scattering (SCS) process from H2 for the case of a single pulse. The thick vertical arrows indicate the absorption and emission of photons of frequency ω1 and ω2, respectively. R is the internuclear distance. The two-shaded areas, blue and purple, show the effective bandwidths after the absorption and the emission of the ω1 and ω2 photons. b Ionization probabilities for the single-pulse case (ω = 20 a.u.). c Idem for the case of two counter-propagating pulses (ω1 = 20 a.u. and ω2 = 15 a.u.). Thick black line: total ionization probability. Blue line: dipole A ⋅ P contribution. Orange line: non-dipole A2 contribution. In both b and c the pulse duration is 200 attoseconds and the peak intensity 1018 W/cm2. Green arrows: pulse polarization direction. Red arrows: pulse incidence direction. Dark orange spheres nuclei. All directions referred to the cartesian x, y, and z axes. Purple wavefronts: attosecond pulse.
Our calculations predict that, for photons of around 20 a.u., SCS probabilities close to the ionization threshold are roughly three orders of magnitude smaller than the one-photon single ionization probabilities leading to electrons of around 20 a.u., and that those probabilities are comparable in magnitude to two-photon single ionization probabilities leading to electrons with about twice this energy. However, the contribution of both one- and two-photon ionization processes close to the ionization threshold is absolutely negligible and lies well below the noise level of the present calculations. Also, standard (unstimulated) Compton scattering is expected to be four orders of magnitude smaller than SCS, as can be shown by using the values of the present pulse parameters in the standard formula that relates both processes. This formula is formally identical to the ratio between the Einstein coefficients for stimulated and spontaneous emission of light (see also Eq. (15) of ref. 8, where c3 should be replaced by c2).
The ionization probabilities for two counter-propagating pulses are shown in Fig. 1c. In this case, both forward and backward Compton scattering processes are possible7. As in the single-pulse case, forward Compton scattering proceeds through absorption of a photon from one of the pulses and stimulated emission from the very same pulse. In contrast, backward Compton scattering requires that a photon is absorbed from one of the pulses and a photon is created by stimulated emission from the counter-propagating pulse. To allow for a clear separation between these two cases, we have chosen different central frequencies for the two pulses: ω1 = 20 a.u. and ω2 = 15 a.u. Figure 1c shows that, as in the single-pulse case, forward Compton scattering leads to a pronounced peak in the vicinity of the ionization threshold, whereas backward Compton scattering leads to a second pronounced peak at electron energies around ω1 − ω2 − Ip ~ 4 a.u., where Ip is the H2 ionization potential. The two peaks are especially apparent in the non-dipole A2 contributions to the ionization probabilities, thus leading to a pronounced oscillation from threshold up to photoelectron energy of ~7 a.u. As shown in Fig. 1c, when the molecule is parallel to the polarization direction, the non-dipole A2 contribution is largely dominant while the opposite is found when the molecule is perpendicular to the polarization direction and parallel to the pulses’ propagation directions. An intermediate situation is observed when the molecule is perpendicular to both the polarization and the photon propagation directions. It is worth noticing that the relative importance of dipole A ⋅ P and non-dipole A2 contributions changes around electron energy of 4 a.u. We also notice that the peaks at the threshold (Fig. 1c) are higher than those of the single-pulse case (Fig. 1b).
Due to the D∞h symmetry of the hydrogen molecule, the following selection rules apply for the dipole A ⋅ P term (two-photon transitions):
and the non-dipole A2 term
As the two terms lead to final continuum states of different inversion symmetry and they add coherently in the transition amplitude when the electron energy is the same in both states, asymmetric electron emission will be possible whenever the corresponding contributions are comparable in magnitude. This will have in turn an important effect on the way the incident photon is scattered.
Molecular-frame photoelectron angular distributions (MFPADs)
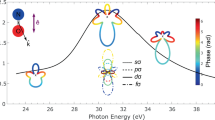
Figure 2 shows the calculated MFPADs for the single-pulse case (same direction for the incoming and the scattered photons) and the three geometrical arrangements considered in Fig. 1b. To ease the comparison with potential experiments in terms of yields, we have integrated over electron energies between 0 and 1.0 a.u., i.e., the region where the cross-section decreases by approximately an order of magnitude from its maximum at zero electron energy. For the three geometrical arrangements, the MFPADs are strongly asymmetric. These asymmetries are also apparent at any given electron energy within the chosen interval (not shown here). When the molecule is parallel to the polarization direction (Fig. 2a) and parallel to the incidence direction (Fig. 2c), forward electron emission is dominant, i.e., electrons escape preferentially in the direction of the incident photon. The opposite is observed when the molecule is perpendicular to both the polarization direction and the light-incidence direction: backward electron emission strongly dominates (Fig. 2b). When the molecule is perpendicular to the polarization direction but parallel to the light-incidence direction, electron emission is more important in the forward direction (Fig. 2c). This behavior is similar to that found in the atomic case7, where forward electron emission along the propagation direction is favored. In the present case, however, the strong variation of the relative phase between the interfering dipole A ⋅ P and non-dipole A2 amplitudes with molecular orientation can also lead to electron emission in the opposite direction. We note that, in the absence of non-dipole effects, the MFPADS would be perfectly symmetric with respect to the inversion center of the molecule.

Green arrows: pulse polarization direction. Red arrows: pulse incidence direction. Dark orange spheres: nuclei. Cuts on the xy, xz, and yx planes are also shown. Molecule is along the z axis. a Polarization along the z axis, incidence direction along the x axis. b Polarization along the x axis, incidence direction along the y axis. c Polarization along the x axis, incidence direction along the z axis.
Figure 3 shows the calculated MFPADs for the case of two counter-propagating pulses (incoming and scattered photons going in opposite directions) and the three geometrical arrangements depicted in Fig. 1c. As in this case we are interested in backward photon scattering (see above), we only show the MFPADs for electron energies ~4 a.u. As can be seen, when the molecule is parallel to the polarization direction (Fig. 3a), there is a slight preference for backward electron emission, in contrast with the single-pulse case (Fig. 2a). When the molecule is perpendicular to both the polarization and the photon incidence directions (Fig. 3b), there is no preference for forwarding or backward electron emission direction, also in contrast with the absolute dominance of backward electron emission in the single-pulse case (Fig. 2b). Even more striking is the fact that, when the molecule is perpendicular to the polarization direction but parallel to the photon incidence directions (Fig. 3c), electrons are preferentially ejected orthogonally to these two directions and in the backward direction. This unusual behavior of the MFPADs is the natural consequence of the interference between the dominant contributions associated with the A2 and A ⋅ P, which, as shown by the selection rules (3–6), involve states of different molecular symmetries. For example, in the last case, the observed MFPAD is the result of the interference between a four-lobed angular distribution resulting from states of \({\,\!}^{1}{{{\Sigma }}}_{g}^{+}\) and 1Δg symmetries (dipole contribution, see Eq. (4)) and a two-lobed angular distribution resulting from a state of \({\,\!}^{1}{{{\Sigma }}}_{u}^{+}\) symmetry (non-dipole contribution, see Eq. (5)).

The central energies are 20 a.u. (blue pulse) and 15 a.u. (purple pulse). Green arrows: pulse polarization direction. Red arrows: pulses incidence direction. Dark orange spheres: nuclei. Cuts on the xy, xz, and yx planes are also shown. Molecule is along the z axis. a Polarization along the z axis, incidence directions along the x axis. b Polarization along the x axis, incidence directions along the y axis. c Polarization along the x axis, incidence directions along the z axis.
Finally, Fig. 4 shows the MFPADs at three different electron energies for two counter-propagating pulses with polarization parallel to the molecular axis. One can see that, as the electron energy increases, electrons go from being preferentially ejected in the forward direction to being ejected in the backward direction, which is the consequence of the change in the relative phase of dipole A ⋅ P and non-dipole A2 contributions in this electron energy range.

Same as Fig. 3a but at different electron energies Ee (a) 1.4 a.u., b 2.5 a.u., and (c) 4.3 a.u. Green arrows: pulse polarization direction. Red arrows: pulses incidence direction. Dark orange spheres: nuclei. Cuts on the xy, xz, and yx planes are also shown. The molecule is along the z axis. Polarization along the z axis, incidence directions along the x axis.
Conclusion
We have calculated MFPADs resulting from SCS of ~500 eV photons by hydrogen molecules. We have found that, by changing molecular orientation with respect to the polarization and photon incidence directions, one can radically change the preferential electron emission direction. In particular, we have shown that it is possible to induce electron emission in either the forward or the backward directions, or in both directions, or even in directions that are orthogonal to the former. This behavior, which can only be observed in molecules, is the consequence of the interference between dipole A ⋅ P and non-dipole A2 interaction contributions, which are magnified in the investigated photon energy range. Experimental realization of this theoretical prediction is nowadays possible by using ultrashort XFEL pulses, which are produced at high enough intensity to compensate for the smallness of the Compton scattering cross-sections at the chosen photon energy. Therefore, this work opens the way to investigations of SCS in molecules, thus providing unforeseen possibilities to manipulate electron dynamics in molecules at its natural time scale.
Extension of the present methodology to include all orders (1/c)n in the description of the light-molecule interaction would allow one to describe even stronger non-dipole effects and, therefore, to provide theoretical support to experiments performed in XFELs at photon energies higher than those used in the present work. Also, the inclusion of nuclear dynamics as described, e.g., in refs. 43,45, would allow one to consider longer pulses and to evaluate vibrationally resolved ionization probabilities, which will be useful to understand the role of nuclear motion in Compton scattering from molecules. Work along these lines is currently in progress. Finally, the use of a fully quantum description of the X-ray/matter interaction as given, e.g., in refs. 49,50, to evaluate the probability of the “unstimulated” Compton scattering process, which may eventually compete with SCS at photon energies higher than those considered in the present work, would also be of great interest, as totally unexplored physical phenomena may be at play in that energy region.
Change history
04 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s42005-021-00784-0
References
Kircher, M. et al. Kinematically complete experimental study of Compton scattering at helium atoms near the threshold. Nat. Phys. 16, 756–760 (2020).
Dörner, R. et al. Cold target recoil ion momentum spectroscopy: a momentum microscope’ to view atomic collision dynamics. Phys. Rep. 330, 95–192 (2000).
Eisenberger, P. & Platzman, P. M. Compton scattering of X rays from bound electrons. Phys. Rev. A 2, 415–423 (1970).
Gavrila, M. Compton scattering by K-shell electrons. I. Nonrelativistic theory with retardation. Phys. Rev. A 6, 1348–1359 (1972).
Doumy, G. et al. Nonlinear atomic response to intense ultrashort X rays. Phys. Rev. Lett. 106, 083002 (2011).
Fuchs, M. et al. Anomalous nonlinear X-ray Compton scattering. Nat. Phys. 11, 964–970 (2015).
Bachau, H., Dondera, M. & Florescu, V. Stimulated compton scattering in two-color ionization of hydrogen with keV electromagnetic fields. Phys. Rev. Lett. 112, 073001 (2014).
Dondera, M., Florescu, V. & Bachau, H. Two-color ionization of hydrogen close to threshold with keV photons. Phys. Rev. A 90, 033423 (2014).
Houamer, S., Chuluunbaatar, O., Volobuev, I. P. & Popov, Y. V. Compton ionization of hydrogen atom near threshold by photons in the energy range of a few kev: nonrelativistic approach. Eur. Phys. J. 74, 81 (2020).
Ackermann, W. et al. Operation of a free-electron laser from the extreme ultraviolet to the water window. Nat. Photonics 1, 336–342 (2007).
Emma, P. et al. First lasing and operation of an ångstrom-wavelength free-electron laser. Nat. Photonics 4, 641–647 (2010).
Jamison, S. X-ray FEL shines brightly. Nat. Photonics 4, 589–591 (2010).
Ishikawa, T. et al. A compact X-ray free-electron laser emitting in the sub-ångström region. Nat. Photonics 6, 540–544 (2012).
Allaria, E. et al. Highly coherent and stable pulses from the FERMI seeded free-electron laser in the extreme ultraviolet. Nat. Photonics 6, 699–704 (2012).
Tschentscher, T. et al. Photon beam transport and scientific instruments at the European XFEL. Appl. Sci. 7, 592 (2017).
Milne, C. et al. SwissFEL: the swiss X-ray free electron laser. Appl. Sci. 7, 720 (2017).
Decking, W. et al. A MHz-repetition-rate hard x-ray free-electron laser driven by a superconducting linear accelerator. Nat. Photonics 14, 391–397 (2020).
Bostedt, C. et al. Linac coherent light source: the first five years. Rev. Mod. Phys. 88, 015007 (2016).
Lutman, A. A. et al. Experimental demonstration of femtosecond two-color x-ray free-electron lasers. Phys. Rev. Lett. 110, 134801 (2013).
De Ninno, G., Mahieu, B., Allaria, E., Giannessi, L. & Spampinati, S. Chirped seeded free-electron lasers: self-standing light sources for two-color pump-probe experiments. Phys. Rev. Lett. 110, 064801 (2013).
Hara, T. et al. Two-colour hard X-ray free-electron laser with wide tunability. Nat. Commun. 4, 2919 (2013).
Serkez, S. et al. Opportunities for two-color experiments in the soft X-ray regime at the european XFEL. Appl. Sci. 10, 2728 (2020).
Callegari, C. et al. Atomic, molecular and optical physics applications of longitudinally coherent and narrow bandwidth free-electron lasers. Phys. Rep. 904, 1–59 (2021).
Eichmann, U. et al. Photon-recoil imaging: expanding the view of nonlinear x-ray physics. Science 369, 1630–1633 (2020).
Kastirke, G. et al. Photoelectron diffraction imaging of a molecular breakup using an x-ray free-electron laser. Phys. Rev. X 10, 021052 (2020).
Chapman, H. Structure determination using x-ray free-electron laser pulses. In Wlodawer, A., Dauter, Z. & Jaskolski, M. (eds.) Protein Crystallography. Methods in Molecular Biology, vol. 1607 (Humana Press, New York, 2017).
Allaria, E. et al. Two-colour pump-probe experiments with a twin-pulse-seed extreme ultraviolet free-electron laser. Nat. Commun. 4, 2476 (2013).
Huang, S. et al. Generating single-spike hard X-ray pulses with nonlinear bunch compression in free-electron lasers. Phys. Rev. Lett. 119, 154801 (2017).
Li, S. et al. Time-resolved pump-probe spectroscopy with spectral domain ghost imaging. Faraday Discuss. 2288 (2021).
Hartmann, N. et al. Attosecond time-energy structure of X-ray free-electron laser pulses. Nat. Photonics 12, 215–220 (2018).
Fukuzawa, H. et al. Real-time observation of X-ray-induced intramolecular and interatomic electronic decay in CH2I2. Nat. Commun. 10, 2186 (2019).
Duris, J. et al. Tunable isolated attosecond X-ray pulses with gigawatt peak power from a free-electron laser. Nat. Photonics 14, 30–36 (2020).
Li, X. et al. Electron-ion coincidence measurements of molecular dynamics with intense x-ray pulses. Sci. Reports 11, 505 (2021).
Samson, J. A. R., He, Z. X., Bartlett, R. J. & Sagurton, M. Direct measurement of He+ ions produced by compton scattering between 2.5 and 5.5 kev. Phys. Rev. Lett. 72, 3329–3331 (1994).
Bachau, H. & Dondera, M. Stimulated Raman scattering in hydrogen by ultrashort laser pulse in the keV regime. EPL (Europhysics Letters) 114, 23001 (2016).
Kircher, M. et al. Recoil-induced asymmetry of nondipole molecular frame photoelectron angular distributions in the hard x-ray regime. Phys. Rev. Lett. 123, 243201 (2019).
Williams, J. B. et al. Imaging polyatomic molecules in three dimensions using molecular frame photoelectron angular distributions. Phys. Rev. Lett. 108, 233002 (2012).
Lucchese, R. R. & Stolow, A. Molecular-frame photoelectron angular distributions. J. Phys. B At. Mol. Opt. Phys. 45, 190201 (2012).
Kastirke, G. et al. Double core-hole generation in o2 molecules using an x-ray free-electron laser: molecular-frame photoelectron angular distributions. Phys. Rev. Lett. 125, 163201 (2020).
Hemsing, E., Stupakov, G., Xiang, D. & Zholents, A. Beam by design: laser manipulation of electrons in modern accelerators. Rev. Mod. Phys. 86, 897–941 (2014).
Weninger, C. et al. Stimulated electronic x-ray raman scattering. Phys. Rev. Lett. 111, 233902 (2013).
O’Neal, J. T. et al. Electronic population transfer via impulsive stimulated x-ray raman scattering with attosecond soft-x-ray pulses. Phys. Rev. Lett. 125, 073203 (2020).
Martín, F. Ionization and dissociation using B-splines: photoionization of the hydrogen molecule. J. Phys. B. At. Mol. Opt. Phys. 32, R197–R231 (1999).
Dondera, M., Florescu, V. & Bachau, H. Stimulated Raman scattering of an ultrashort XUV radiation pulse by a hydrogen atom. Phys. Rev. A 95, 023417 (2017).
Palacios, A., Sanz-Vicario, J. L. & Martín, F. Theoretical methods for attosecond electron and nuclear dynamics: applications to the H2 molecule. J. Phys. B: At. Mol. Opt. Phys. 48, 242001 (2015).
Waitz, M. et al. Imaging the square of the correlated two-electron wave function of a hydrogen molecule. Nat. Commun. 8, 2266 (2017).
Palacios, A., Barmaki, S., Bachau, H. & Martín, F. Two-photon ionization of H2 by short laser pulses. Phys. Rev. A 71, 063405 (2005).
Sanz-Vicario, J. L., Bachau, H. & Martín, F. Time-dependent theoretical description of molecular autoionization produced by femtosecond xuv laser pulses. Phys. Rev. A 73, 033410 (2006).
Henriksen, N. E. & Møller, K. B. On the theory of time-resolved x-ray diffraction. J. Phys. Chem. B 112, 558–567 (2008).
Dixit, G., Vendrell, O. & Santra, R. Imaging electronic quantum motion with light. Proc. Natl. Acad. Sci. 109, 11636–11640 (2012).
Acknowledgements
All calculations were performed at the Mare Nostrum Supercomputer of the Red Española de Supercomputación (BSC-RES), the Centro de Computación Científica de la Universidad Autónoma de Madrid (CCC-UAM), and Mésocentre de Calcul Intensif Aquitain (MCIA). Work supported by the European COST Action AttoChem, the Spanish MICINN projects PID2019-105458RB-I00, the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2016-0686) and the “María de Maeztu” Programme for Units of Excellence in R&D (CEX2018-000805-M), the Comunidad de Madrid Synergy Grant FULMATEN, and the French National Research Agency (ANR) in the framework of “the Investments for the future” Programme IdEx Bordeaux - LAPHIA (ANR-10-IDEX-03-02).
Author information
Authors and Affiliations
Contributions
H.B., A.P., and F.M. conceived the project. A.S. performed the theoretical calculations and implemented the necessary tools to analyze the results. A.P., F.C., H.B., and F.M. supervised the project. A.S., A.P., and F.M. built the original version of the computer code. F.M. wrote the first draft of the manuscript. All authors analyzed the results and contributed to the preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
There are no competing interests.
Additional information
Peer review information Communications Physics thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sopena, A., Palacios, A., Catoire, F. et al. Angle-dependent interferences in electron emission accompanying stimulated Compton scattering from molecules. Commun Phys 4, 253 (2021). https://doi.org/10.1038/s42005-021-00749-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-021-00749-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
