
Abstract
Accurate detection and risk stratification of latent tuberculosis infection (LTBI) remains a major clinical and public health problem. We hypothesize that multiparameter strategies that probe immune responses to Mycobacterium tuberculosis can provide new diagnostic insights into not only the status of LTBI infection, but also the risk of reactivation. After the initial proof-of-concept study, we developed a 13-plex immunoassay panel to profile cytokine release from peripheral blood mononuclear cells stimulated separately with Mtb-relevant and non-specific antigens to identify putative biomarker signatures. We sequentially enrolled 65 subjects with various risk of TB exposure, including 32 subjects with diagnosis of LTBI. Random Forest feature selection and statistical data reduction methods were applied to determine cytokine levels across different normalized stimulation conditions. Receiver Operator Characteristic (ROC) analysis for full and reduced feature sets revealed differences in biomarkers signatures for LTBI status and reactivation risk designations. The reduced set for increased risk included IP-10, IL-2, IFN-γ, TNF-α, IL-15, IL-17, CCL3, and CCL8 under varying normalized stimulation conditions. ROC curves determined predictive accuracies of > 80% for both LTBI diagnosis and increased risk designations. Our study findings suggest that a multiparameter diagnostic approach to detect normalized cytokine biomarker signatures might improve risk stratification in LTBI.
Similar content being viewed by others


Introduction
Tuberculosis (TB) is a pervasive and devastating infectious disease that led to 1.2 million deaths in 20191. Beyond the acute morbidity of the disease, it also can manifest as an asymptomatic infection known as latent tuberculosis (LTBI), which greatly complicates disease management. Approximately 1.7 billion people are estimated to have this quiescent infection state1,2,3. Among this population, 5–10% are estimated to develop an active, transmissible, and potentially lethal TB infection4,5,6. LTBI is treatable with prolonged antibiotic regimens, but potential drug-related toxicities and treatment non-adherence issues obviate the need to identify patients most likely to benefit from these therapies. To properly diagnose individuals with LTBI, and more specifically those with high reactivation potential, improved diagnostic tools are needed. While current assays for TB, including the Tuberculin Skin Test (TST) and Interferon-Gamma Release Assays (IGRAs), reveal previous exposure to TB, they offer limited benefit for LTBI diagnosis and reactivation risk stratification7. There is a growing consensus that this latent infection and the conditions that foster reactivation include complex changes in the immunological landscape that may be independent of the pathogen itself8,9,10,11,12,13,14. Therefore, multiparameter strategies that probe immune (dys)regulation in response to TB-specific and non-specific antigen challenge may provide new diagnostic insights into not only the status of LTBI infection, but also the risk of reactivation.
Previously, our groups described an approach to multiplexed cytokine profiling of supernatants from stimulated peripheral blood mononuclear cells (PBMCs) using arrays of silicon photonic microring resonators15. This workflow involved five different TB-specific and non-specific stimulation conditions, and the quantification of seven cytokines. Using a precision normalization approach to correct for individual patient heterogeneity and a bioinformatic feature selection approach, we identified multi-biomarker signatures that preliminarily correlated with LTBI status and elevated reactivation potential. This study suggested relevant biomarkers beyond interferon-gamma (IFN-γ), which is the cytokine detected in IGRAs, and also highlighted the importance of precision normalization using non-TB-related antigen challenges as a way of accounting for differences in basal immune response. However, this was only a first proof-of-concept cohort study towards the development of a new LTBI detection paradigm as it focused on a limited number of cytokines and a relatively small population of research subjects.
Building upon this previous cohort study, we have now expanded to a larger panel of cytokines (13), again detected using silicon photonic microring sensor arrays, and enrolled a larger research cohort (65 subjects, 75 unique samples). We also have added a new antigen condition with the MTB300 reagent, which is a comprehensive “megapool” of Mycobacterium tuberculosis (Mtb) peptides that captures a large fraction of the Mtb-specific T cells16. Cytokine concentrations were determined for each stimulation condition and normalized by pairwise subtraction of levels from other stimulation conditions. The resulting normalized cytokine levels were then subjected to Random Forest feature selection to identify biomarker signatures correlating with LTBI + status and High Risk of reactivation (the entire workflow is illustrated in Fig. 1). Statistically-driven thresholding was then applied to identify the most relevant biomarkers having the highest predictive accuracy for these clinical designations. Receiver-Operator Characteristic (ROC) curves were generated for both full and reduced data sets with Area-Under-the-Curve (AUC) analyses revealing predictive accuracies exceeding 80% for reduced biomarker signatures. These studies suggest that multiplexed analyses and precision cytokine normalization can generate highly predictive signatures for LTBI status and reactivation potential that may find utility in clinical management of patients with LTBI through a personalized medicine approach.

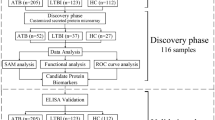
Workflow for LTBI supernatant sample analysis. (A) Subject PBMCs are stimulated under multiple on- and off-target conditions. (B) Samples are analyzed using the Genalyte Matchbox system, which uses plug-and-play chip and device interfaces to measure cytokine concentrations quickly and reproducibly in a multiplexed assay format. The resonance shift output is recorded and converted to concentrations based on individual cytokine calibrations in the sample matrix. (C) Random Forest bioinformatics determine what clinical features are essential for categorical distinctions and predictive accuracy based on variable importance metrics, with statistical data reduction methods employed to identify biomarker signatures most highly correlated with given clinical determinants.
Methods
Reagents and buffers
Reagents, including Dulbecco’s phosphate buffered saline (PBS), bovine serum albumin (BSA), (3-Aminopropyl)triethoxysilane, glycerol, bis(sulfosuccinimidyl)suberate, starting block blocking buffer, Pierce high sensitivity streptavidin-HRP (SA-HRP), 4-chloronaphthol (4-CN), and Drycoat assay stabilizer were purchased from commercial vendors as listed in Table S1. Vendors and catalog numbers for antibodies against all cytokines and the mouse IgG isotype control are summarized in Table S2. Running buffer for all assays was 0.5% BSA in 1X PBS, pH 7.4.
Cell culture and antigen stimulations
Cell culture and antigen stimulation methods have been described previously15,17. Briefly, PBMCs were separated by from whole blood by Ficoll separation and the pellets frozen with 10% DMSO in cell media in liquid nitrogen. PBMC pellets were thawed (viability ≥ 85%) and 1 million cells per mL were stimulated for 40–48 h with either TB-relevant (PPD [10 µg/mL], CFP-10 [4 µg/mL]/ESAT-6 [2 µg/mL], or MTB300 [1 µg/mL]) or off-target (Media—negative control, Candida—off-target control [2 µg/mL], anti-CD3—positive control [200 ng/mL]) antigens17. Supernatants from stimulated PBMCs were stored at − 80 °C and shipped on dry ice for cytokine measurements using the 13-plex antigen immunoassay on the microring resonators. Samples were thawed and vortexed prior to loading into a 96-well plate for microring assays. Each sample was analyzed undiluted and after a tenfold dilution in running buffer.
Clinical category determination
This study was approved by the Mayo Clinic Institutional Review Board and Olmsted County Public Health Services (reference number: 09-003253). All study participants signed an informed written consent and were enrolled in Rochester, MN between July 2017 and December 2018. Sample collection and all experiments were performed in accordance with relevant guidelines and regulations. Study subjects included unexposed individuals and subjects with various risk for TB infection, including untreated LTBI patients and patients with having had LTBI therapy and thus at low risk of reactivation (Table 1). Risk factors for TB infection, TB progression, and/or TB reactivation were extracted through a validated questionnaire and review of medical records as previously described15,18. LTBI diagnoses were made per the Center for Disease Control and Prevention (CDC) criteria and based on TB risk factors, and by prior TST and QuantiFERON-TB Gold In-Tube (QFT; Qiagen, Germantown, MD) results19. A modified multifactorial predictive modeling platform (i.e. ‘Online TST/IGRA interpreter’), adjusted by LTBI treatment effect, was also applied to estimate the cumulative risk of TB reactivation in all subjects as previously described17,20.
Multiplexed immunoassay instrumentation and assay design
Microring immunoassays were performed on the Maverick Matchbox system (Genalyte, Inc., San Diego, CA) as previously described21. This platform utilizes injection-molded microfluidic devices to introduce fully automated flow for all assay steps across functionalized sensor arrays. These devices are disposable, to ensure no contamination between assays22,23. Microring arrays were batch functionalized via spotting to yield a 13-plex array of capture antibodies covalently immobilized on sensor substrates. After flowing the sample across the chip, a cocktail containing all the tracer antibodies was flowed across the array followed by signal enhancement reagents. Antibody capture and tracer concentrations used are listed in Table S3. Shifts in resonance wavelength from the signal enhancement step are directly correlated to the concentration of target analytes in solution. Immunoassays were performed with a consistent 30 μl/min flow rate for all steps. There was an initial rinse of 5 min with the running buffer to ensure equilibration of the chip prior to sample analysis. The assay included steps as follows: (1) running buffer (2 min); (2) sample (7 min); (3) running buffer rinse (2 min); (4) biotinylated tracer antibodies (7 min); (5) running buffer rinse (2 min); (6) SA-HRP (7 min); (7) running buffer rinse (2 min); (8) 4-CN (7 min); (9) running buffer rinse (2 min). The total assay time was 38 min. Figure S1 shows a real-time trace of resonance wavelength shifts during a representative multiplexed immunoassay.
Calibrations and sample analyses
Immunoassays were simultaneously calibrated for all antigens in a multiplexed format. Serial dilutions from a saturating antigen concentration for each multiplexed immunoassay yielded eight-point calibrations relating relative resonance shifts to standard concentrations. To quantify relative shifts, the signal during the buffer rinse before the assay enhancement step (t = 29 min) was subtracted from the final assay rinse step (t = 38 min). Net resonance wavelength shifts (\(\Delta pm\)) were plotted as a function of standard concentration and fit to a four-parameter logistic function as described previously21 via the following equation:
where \(A_{1}\) is the lower resonance shift bound, \(A_{2}\) is the upper resonance shift bound, \(\left[ {antigen} \right]_{0.5}\) is the concentration yielding 50% of maximum signal, and \(p\) is the power parameter affecting the slope at \(\left[ {antigen} \right]_{0.5}\). Limits of detection (LOD) and quantification (LOQ) were defined as the blank signal plus 3 times and 10 times the standard deviation of the blank, respectively. Each calibration was performed in triplicate for each sample dilution (Fig. S2) as measured with 4 sensors per technical replicate.
Samples were analyzed undiluted (1X) and diluted (0.1X) into running buffer. Cytokine concentrations within supernatant samples were determined from corresponding serum calibrations (10% and 1% serum, respectively, matching the serum content in supernatant samples). Final concentrations were measured using the most appropriate dilution/calibration as determined by how close the value came to the midpoint of the calibration. Precision normalization was achieved by separately subtracting the control or off-target cytokine concentration from that measured at the other stimulation conditions.
Random forest and ROC curve analyses
Random Forest methods were utilized to determine the biomarker features associated with both LTBI diagnosis (“LTBI + ”) and increased risk of reactivation (“High Risk”) clinical designations24. Random Forest is an ensemble classification algorithm that can detect nonlinear effects of covariate features. Moreover, it provides a ranking of importance of each covariate, while the importance is identified by their effect on classification of either LTBI or risk of reactivation. Using these randomized decision tree outputs, a ROC curve can be established that indicates the predictive accuracy the variables hold for a given classification.
Feature importance analysis was done for all variables available (143 total), and ROC curves were produced. Utilizing the important features from those analyses, a secondary Random Forest algorithm was produced for further enhancement of predictive accuracy by removing unimportant or noisy features. This was done identically for both LTBI + and High Risk clinical designations. Mann–Whitney plots were utilized to evaluate each important variable in the signature. All informatics and plotting were performed using R coding language25,26.
Results
Study subjects
We sequentially enrolled a total of 65 subjects, including 32 subjects with diagnosis of LTBI by QFT and/or TST results and 5 immunosuppressed patients for various medical conditions. The total sample set included 75 samples, encompassing 65 unique subjects and 10 additional time points separated by 5–11 months in testing, representing unique samples (Table 1). All HIV-tested subjects were non-reactive by ELISA (45 out of 65). The majority of study participants were health care workers (73%) with various risks of TB exposure and 10 (15.4%) had prior history of BCG vaccination. Twenty-three subjects with negative QFT results and a predicted cumulative TB risk of zero were included as unexposed control subjects (Table 1). Eight individuals who previously received treatment for LTBI and were considered at low risk for TB reactivation. There were no significant age differences across the study clinical designations.
Full versus reduced random forest feature selection
The initial Random Forest analysis was performed using all possible normalized features from the full dataset (13 cytokines × 11 normalized, pairwise conditions), identifying 143 features viable for the LTBI + clinical designation. Based on all possible features, a ROC curve for LTBI + was produced (Fig. 2A), yielding an AUC of 0.767 (76.7% predictive value). From this full feature analysis, a reduced random forest validation containing all the important features from the full analysis was created using the statistical variance of each feature and establishing a threshold based on predictive accuracy improvement. Therefore, the 19 identified features after data reduction, which span a variety of cytokines and normalized conditions, can be considered as the most relevant to the ROC curve’s predictive power (Fig. 2B). A ROC curve constructed using only these reduced features yielded an AUC of 0.874 (Fig. 2C). Mann–Whitney analyses of the statistical significance of variables included in the reduced analysis are detailed in Table S4.

Comparison of (A) full random forest feature and (B) the threshold-based reduced random forest feature analysis for the LTBI + clinical category. Features for the reduced analysis are determined by Variable Importance (VIMP) metrics. (C) ROC Curves for full and reduced analysis represent the predictive power of each method, with AUC values corresponding to the percent predictive accuracy. Notably, the reduced biomarker set offers improved predictive accuracy. Abbreviations—MED (cell media), CAN (Candida), CD3 (anti-CD3), PPD (purified protein derivative), CE (CFP-10/ESAT-6), MTB (MTB300). Normalized conditions are denoted as Condition 1 minus Condition 2 (i.e. PPD-MED is the PPD condition minus the negative control cell media condition).
Importance of MTB300 stimulation condition
To assess the added benefit of the MTB300 stimulation condition, which specifically targets a large fraction of Mtb-specific CD4+ and CD8+ T cell populations, separate Random Forest feature selection and data reduction algorithms were performed for LTBI + in the absence of any normalized conditions that included the MTB300 stimulation (Fig. 3A). A quantitative comparison between the previous reduced feature set for LTBI and the further reduced analysis without MTB300 shows a loss of 5.3% in prediction accuracy (AUC changed from 0.874 to 0.821), indicating a valuable role for MTB300 in improving LTBI diagnostic utility (Fig. 3B).

(A) Reduced random forest feature analysis for the LTBI + clinical designation when the MTB stimulation is removed. (B) ROC curve comparison of LTBI + reduced random forest with and without MTB stimulation condition. This indicates an improved predictive accuracy through the inclusion of the MTB stimulation condition.
Stratification of subjects based upon high risk of reactivation
Reduced Random Forest analysis was performed using the entire normalized dataset as input for the High Risk of reactivation clinical designation. Though this subpopulation is smaller than the LTBI + pool, it is still large enough for robust feature identification. The full data set analysis for High Risk used all 143 features with a resulting AUC of 0.715 (Fig. 4A). Upon performing the statistical feature reduction, 14 features were identified as most relevant, and these reduced features yielded a predictive accuracy of 0.855 (Fig. 4B, C). Mann–Whitney analyses of the statistical significance of these reduced biomarkers is detailed in Table S5.

Comparison of (A) full random forest feature and (B) the threshold-based reduced random forest feature analysis for the High Risk clinical designation. (C) ROC Curves for full and reduced analysis show an increase in predictive accuracy for the reduced feature set.
Discussion
LTBI is a persistent and quiescent infection that has proven difficult to identify accurately using conventional diagnostic methods. The added stratification of progression and/or reactivation potential has further been a diagnostic challenge that limits the ability to robustly identify patients most likely to benefit the most from antibiotic treatment. Utilizing our 13-target multiplexed cytokine panel and powerful bioinformatic approaches, we have identified multi-biomarker signatures that show strong correlations to clinical designations of LTBI + and High Risk of reactivation with an approach that only uses input biomarker levels to achieve these diagnoses.
Based upon our previous study, we found that precision normalization revealed a richer biomarker signature compared to feature selection using non-normalized cytokine levels within stimulated PBMC supernatants. We attributed this to elimination of patient-to-patient heterogeneities in basal immune responses, which were corrected by subtracting non-TB control levels. Therefore, we utilized normalized cytokine levels obtained by subtracting control and off-target antigen stimulation conditions from other supernatant solutions. Using these normalized values as input, iterative feature selection methods were applied as described to identify biomarkers that first correlated with the LTBI + designation, as determined via extensive clinical chart review. Using all 143 possible features from the full dataset (Fig. 2A), a ROC curve was generated and found to have an AUC of 0.767, or a 76.7% predictive accuracy (Fig. 2C). Realizing that some features might have more overall significance than others, a statistical data reduction routine was performed, identifying 19 features that most strongly correlated with LTBI + (Fig. 2B). Biomarkers in the reduced set included IL-2, IFN-γ, IP-10, CCL8, CCL2, IL-6, and CCL3 under varying normalized stimulation conditions. The ROC/AUC analysis using just the reduced feature showed a predictive accuracy of 87.4%, which was a 10.7% improvement over the full feature set (Fig. 2C). We hypothesize that some of the features in the full data set feature reduction, while weakly correlated with LTBI + , contained higher levels of biological variance that reduce the overall predictive potential across the diverse LTBI + cohort. Application of this thresholding approach to data reduction focused the biomarker signature on those features most relevant, thereby increasing the predictive accuracy of the ROC curve and diagnostic efficacy. For future studies, this realization is also important as it will allow for the development of more focused and simplified cytokine panels that do not require non-essential biomarkers.
Beyond the inclusion of more cytokines in this updated panel, we also included the MTB300 stimulation condition, so as to probe the relative value of this stimulation condition in more accurately diagnosing LTBI. MTB300 is a mixture of 300 Mtb-derived T cell epitopes that specifically targets a large fraction of Mtb-specific CD4+ and CD8+ T cells16,27. Both Purified Protein Derivative (PPD) and CFP-10/ESAT-6 (CE) have traditionally been used in TST and QFT testing, respectively. While these stimulations can elucidate an immune response that is indicative of tuberculosis exposure, they are potentially insufficient to differentiate LTBI or reactivation risk. In addition, the majority of the human response to peptide antigens from Mtb is not contained in the peptide mixtures of IGRAs16. In this context, the use of the MTB300 “megapool” of peptides in our immune profiling method may theoretically confer a better sensitivity over IGRA methods by targeting a larger fraction of the Mtb-specific T cells. Importantly, MTB300 contains 300 primarily MHC class II restricted T cell epitopes from 90 different antigens in Mtb. While these epitopes are derived from Mtb many of these epitopes are also conserved in the M. bovis bacillus Calmette-Guérin (BCG) and also non-tuberculous mycobacteria. To verify that the new stimulation condition adds value to our multiplexed cytokine profiling approach, we performed identical feature selection and data reduction methods omitting all normalized features that contained the MTB condition. Focusing just on the reduced feature sets for LTBI + for comparison (Fig. 3), we found that AUCs fell by 5.3% upon the removal of the MTB condition, indicating the importance of MTB300 for improved diagnostic accuracy.
Similarly to the LTBI + designation, we also performed identical feature selection of both the full normalized feature set and after data reduction for the High Risk clinical designation (Fig. 4). Again, the reduced data set yields an improved predictive value (85.5% reduced vs. 71.5% full). Additionally, we find a similar number of relevant biomarker features for High Risk and LTBI + designations, with IFN-γ, IP-10, IL-2, IL-6, CCL3, and CCL8 appearing in both designations. These targets are important in T cell recruitment, granuloma formation, and inflammatory regulation28,29,30,31,32, and so the overlapping relevance for these biomarkers is not unsurprising9,10,33. Interestingly, we also find some markers that are unique between the designations. Specifically, the chemotactic marker CCL2, which is induced by tissue injury or infection34, was identified as relevant for LTBI + , but not for High Risk. Conversely, the cytokines TNF-α, IL-17, and IL-15 were found to be uniquely relevant for the High Risk designation35,36,37,38. Given that LTBI reactivation is thought to occur due to global changes to host immune regulation, it is intriguing that cytokines not typically associated with the acute TB infection response to have diagnostic utility in reactivation risk stratification. As a corollary, patients on anti-TNF-α therapies are known to have higher risk of reactivation, which suggests the potential for diagnostic monitoring of multiple immune regulatory factors when surveilling for reactivation risk.
Overall, we found that this multiplexed, precision normalization approach to diagnosing LTBI and stratifying high reactivation potential resulted in multi-biomarker signatures with AUCs in excess of 0.80 for reduced feature sets. This study highlights the critical value in moving beyond single biomarker-based methods, such as IGRAs that just analyze for IFN-γ. Furthermore, we found that MTB300 provides a measurable improvement in the predictive accuracy of the LTBI + diagnostic signature. It also validates our platform as a potential point-of-care system with fast response times for clinical implementation.
Our study does have some potential limitations. Specifically, there is no diagnostic gold standard for LTBI, and available diagnostic tests are imperfect. However, our study subjects were carefully selected to minimize heterogeneity in the study groups and to align with current clinical diagnostic standards18. In addition, most of our study cohort included non-HIV immunocompetent individuals, which could limit the applicability of our study findings to immunosuppressed subjects and people with HIV infection. Another potential limitation is the use of the ‘Online TST/IGRA interpreter,’ which has not been prospectively validated. However, we decided to use this predictive tool for research purposes in absence of any other unbiased clinical tool that theoretically quantifies cumulative risk of TB reactivation in LTBI using TST and/or IGRA results plus evaluation of an individual’s most relevant epidemiologic and clinical characteristics20. Moreover, the annual risk of infection estimated by the ‘Online TST/IGRA interpreter’ is based on long-term longitudinal data of TB reactivation39. We also understand that without a direct comparison to a cohort of active TB patients, we are missing potential classifiers to understand progression to an active TB disease. While we enrolled some patients with active TB, there were too few to statistically compare with our High Risk designation signature, and we plan to further study this important cohort in the future. Lastly, nine study subjects participated twice several months after their initial study participation; however, we decided to include their study data since some of them had changes their follow-up QFT results, and thus their LTBI diagnostic designation. These immune profiling changes probably represent the dynamic nature of LTBI in some of these subjects, which we wanted to also capture during this study.
Conclusion
Using this expanded 13-plex cytokine immunoassay biosensor panel, we have demonstrated the ability to identify complex biomarker signatures for LTBI status and to elucidate targets of interest for reactivation risk assessment. Our bioinformatics approach using Random Forest feature selection has shown excellent utility in determining important biomarkers and normalized conditions vital for clinical discrimination between subjects. Furthermore, the feature reduction leads to an improved predictive accuracy and the potential to optimize the multiplexed panel to its most essential components for simplified assay development. We found multiple markers that are essential for both LTBI and reactivation risk, including IFN-γ, IP-10, IL-2, IL-6, CCL3, and CCL8, which represents a broader immunological profile than single target assays. Conversely, distinctive targets such as CCL2 reveal subjects at higher risk of reactivation from the greater LTBI + cohort. We also revealed the value of the peptide pool MTB300 for future stimulated cell-based analyses through data reduction methods. Predictive accuracies for both designations were above 80%, which indicates the potential for improved patient monitoring and clinical care. This biosensor technique, combined with robust bioinformatics, supports this unique strategy for biomarker-only signatures yielding reliable evidence-based clinical decisions for LTBI and the reactivation risk.
References
WHO. Global Tuberculosis Report 2020 (WHO, 2020).
Ernst, J. D. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 12, 581–591 (2012).
Houben, R. M. G. J. & Dodd, P. J. The global burden of latent tuberculosis infection: A re-estimation using mathematical modelling. PLoS Med. 13, e1002152 (2016).
Esmail, H., Barry, C. E., Young, D. B. & Wilkinson, R. J. The ongoing challenge of latent tuberculosis. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 369, 20130437 (2014).
Salgame, P., Geadas, C., Collins, L., Jones-López, E. & Ellner, J. J. Latent tuberculosis infection—Revisiting and revising concepts. Tuberculosis (Edinb.) 95, 373–384 (2015).
Mack, U. et al. LTBI: latent tuberculosis infection or lasting immune responses to M. tuberculosis? A TBNET consensus statement. Eur. Respir. J. 33, 956–973 (2009).
Rangaka, M. X. et al. Predictive value of interferon-γ release assays for incident active tuberculosis: A systematic review and meta-analysis. Lancet Infect. Dis. 12, 45–55 (2012).
Lu, L. L. et al. IFN-γ-independent immune markers of Mycobacterium tuberculosis exposure. Nat. Med. 25, 977–987 (2019).
Porter, B. W. & Venkatappa, T. K. Uncloaking an ancient adversary: Can pathogen biomarker elicitors play a role in confirming extrapulmonary TB and latent TB infection?. Tuberculosis 113, 30–37 (2018).
Essone, P. N. et al. M. tuberculosis infection and antigen specific cytokine response in healthcare workers frequently exposed to tuberculosis. Sci. Rep. 9, 1–13 (2019).
Kim, S. Y. et al. The responses of multiple cytokines following incubation of whole blood from TB patients, latently infected individuals and controls with the TB antigens ESAT-6, CFP-10 and TB7.7. Scand. J. Immunol. 76, 580–586 (2012).
Kellar, K. L. et al. Multiple cytokines are released when blood from patients with tuberculosis is stimulated with Mycobacterium tuberculosis antigens. PLoS ONE 6, e26545 (2011).
Chegou, N. N., Black, G. F., Kidd, M., van Helden, P. D. & Walzl, G. Host markers in Quantiferon supernatants differentiate active TB from latent TB infection: Preliminary report. BMC Pulm. Med. 9, 21 (2009).
Wang, X. et al. Diagnostic performance of multiplex cytokine and chemokine assay for tuberculosis. Tuberculosis (Edinb.) 92, 513–520 (2012).
Robison, H. M. et al. Precision immunoprofiling to reveal diagnostic signatures for latent tuberculosis infection and reactivation risk stratification. Integr. Biol. (Camb.) 11, 16–25 (2019).
Arlehamn, C. S. L. et al. A quantitative analysis of complexity of human pathogen-specific CD4 T cell responses in healthy M. tuberculosis infected South Africans. PLoS Pathog. 12, e1005760 (2016).
Escalante, P. et al. Combinatorial immunoprofiling in latent tuberculosis infection. Toward better risk stratification. Am. J. Respir. Crit. Care Med. 192, 605–617 (2015).
El Sahly, H. M. et al. Epidemiologic differences between United States—And foreign-born tuberculosis patients in Houston, Texas. J. Infect. Dis. 183, 461–468 (2001).
Lewinsohn, D. M. et al. Official American thoracic society/infectious diseases society of America/centers for disease control and prevention clinical practice guidelines: Diagnosis of tuberculosis in adults and children. Clin. Infect. Dis. 64, 111–115 (2017).
Menzies, D., Gardiner, G., Farhat, M., Greenaway, C. & Pai, M. Thinking in three dimensions: A web-based algorithm to aid the interpretation of tuberculin skin test results. Int. J. Tuberc. Lung Dis. 12, 498–505 (2008).
Robison, H. M. & Bailey, R. C. A guide to quantitative biomarker assay development using whispering gallery mode biosensors. Curr. Protoc. Chem. Biol. 9, 158–173 (2017).
Mudumba, S. et al. Photonic ring resonance is a versatile platform for performing multiplex immunoassays in real time. J. Immunol. Methods 448, 34–43 (2017).
Miyara, M. et al. Detection in whole blood of autoantibodies for the diagnosis of connective tissue diseases in near patient testing condition. PLoS ONE 13, e0202736 (2018).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
R Core Team. R: A Language and Environment for Statistical Computing (R Core Team, 2013).
Ishwaran, H. & Kogalur, U. B. Package ‘randomForestSRC’ (2020).
Yang, J. D. et al. Mycobacterium tuberculosis-specific CD4+ and CD8+ T cells differ in their capacity to recognize infected macrophages. PLoS Pathog. 14, e1007060 (2018).
Ruhwald, M., Bjerregaard-Andersen, M., Rabna, P., Eugen-Olsen, J. & Ravn, P. IP-10, MCP-1, MCP-2, MCP-3, and IL-1RA hold promise as biomarkers for infection with M. tuberculosis in a whole blood based T-cell assay. BMC Res. Notes 2, 19 (2009).
Martinez, A. N., Mehra, S. & Kaushal, D. Role of interleukin 6 in innate immunity to Mycobacterium tuberculosis infection. J. Infect. Dis. 207, 1253–1261 (2013).
Singh, B., Chitra, J. & Selvaraj, P. CCL2, CCL3 and CCL4 gene polymorphisms in pulmonary tuberculosis patients of South India. Int. J. Immunogenet. 41, 98–104 (2014).
Tang, N.L.-S. et al. Genetic association between a chemokine gene CXCL-10 (IP-10, interferon gamma inducible protein 10) and susceptibility to tuberculosis. Clin. Chim. Acta 406, 98–102 (2009).
Millington, K. A. et al. Dynamic relationship between IFN-γ and IL-2 profile of Mycobacterium tuberculosis-specific T cells and antigen load. J. Immunol. 178, 5217–5226 (2007).
Ahmed, M. et al. Immune correlates of tuberculosis disease and risk translate across species. Sci. Transl. Med. 12, eaay0233 (2020).
Hasan, Z. et al. CCL2 responses to Mycobacterium tuberculosis are associated with disease severity in tuberculosis. PLoS ONE 4, e8459 (2009).
Harari, A. et al. Dominant TNF-α+ Mycobacterium tuberculosis-specific CD4+ T cell responses discriminate between latent infection and active disease. Nat. Med. 17, 372–376 (2011).
Yoshida, Y. O. et al. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J. Immunol. 184, 4414–4422 (2010).
Khader, S. A. et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377 (2007).
Umemura, M., Nishimura, H., Hirose, K., Matsuguchi, T. & Yoshikai, Y. Overexpression of IL-15 in vivo enhances protection against Mycobacterium bovis bacillus Calmette-Guérin infection via augmentation of NK and T cytotoxic 1 responses. J. Immunol. 167, 946–956 (2001).
Styblo, K. The relationship between the risk of tuberculosis infection and the risk of developing infectious tuberculosis. Bull. Int. Union Tuberc. 60, 117–119 (1985).
Acknowledgements
We thank the study subjects for their generous participation. We thank the staff and colleagues of the Olmsted Public Health Department Tuberculosis Clinic, and the Lanmark Pulmonary Research Unit for their diligent support and assistance. We appreciate the staff and colleagues of the Mayo Mycobacterial and Bronchiectasis Clinic, and the Mayo Clinic Infectious Disease Serology Laboratory for their valuable support.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health [AI141591 to R.C.B. and P.E.]. Part of this Project was also supported by Grant Number UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS). This paper’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health, Mayo Clinic, or any organization. No other financial or material support for this work was provided to the authors and participants.
Ethics declarations
Competing interests
R.C.B. has a minor financial interest in Genalyte, Inc. P.E. and T.P., and their institution have filed two patent applications related to immunodiagnostic laboratory methodologies for latent tuberculosis infection (Patent numbers: 9678071 and 10401360), which are not included in this manuscript. To date, there has been no income or royalties associated with those filed patent applications. PE participated in a short-term advisory scientific board for DiaSorin Molecular, which was outside the scope of the submitted manuscript, and honorarium was paid to Mayo Clinic. P.E., and T.P. have no other conflicts to declare. H.M.R., C.A.C., H.Z., C.L.E., E.T, C.S.L.A., A.S., C.B., M.W., and R.Z. have no conflicts to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Robison, H.M., Chapman, C.A., Zhou, H. et al. Risk assessment of latent tuberculosis infection through a multiplexed cytokine biosensor assay and machine learning feature selection. Sci Rep 11, 20544 (2021). https://doi.org/10.1038/s41598-021-99754-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-99754-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
