
Abstract
This multicenter phase-II trial aimed to investigate the efficacy, safety, and predictive biomarkers of toripalimab plus chemotherapy as second-line treatment in patients with EGFR-mutant-advanced NSCLC. Patients who failed from first-line EGFR-TKIs and did not harbor T790M mutation were enrolled. Toripalimab plus carboplatin and pemetrexed were administrated every three weeks for up to six cycles, followed by the maintenance of toripalimab and pemetrexed. The primary endpoint was objective-response rate (ORR). Integrated biomarker analysis of PD-L1 expression, tumor mutational burden (TMB), CD8 + tumor-infiltrating lymphocyte (TIL) density, whole-exome, and transcriptome sequencing on tumor biopsies were also conducted. Forty patients were enrolled with an overall ORR of 50.0% and disease-control rate (DCR) of 87.5%. The median progression free survival (PFS) and overall survival were 7.0 and 23.5 months, respectively. The most common treatment-related adverse effects were leukopenia, neutropenia, anemia, ALT/AST elevation, and nausea. Biomarker analysis showed that none of PD-L1 expression, TMB level, and CD8 + TIL density could serve as a predictive biomarker. Integrated analysis of whole-exome and transcriptome sequencing data revealed that patients with DSPP mutation had a decreased M2 macrophage infiltration and associated with longer PFS than those of wild type. Toripalimab plus chemotherapy showed a promising anti-tumor activity with acceptable safety profiles as the second-line setting in patients with EGFR-mutant NSCLC. DSPP mutation might serve as a potential biomarker for this combination. A phase-III trial to compare toripalimab versus placebo in combination with chemotherapy in this setting is ongoing (NCT03924050).
Similar content being viewed by others


Introduction
The therapeutic landscape has dramatically shifted during the past decade for advanced non-small-cell lung cancer (NSCLC) patients with EGFR-sensitizing mutations.1,2 First-line setting with EGFR-TKIs, such as erlotinib, gefitinib, icotinib, afatinib, dacomitinib, and osimertinib, has resulted in significantly longer progression-free survival (PFS) than the previous standard of care.3,4,5,6,7,8,9 However, after failure of first-line EGFR-TKI therapy, PFS with second or subsequent lines of treatment is disappointing with 4.4–5.4 months in EGFR T790M-negative group that received chemotherapy.10,11,12 Even though those with EGFR T790M mutations received osimertinib, resistance is also inevitable and subsequent treatments often show very limited clinical efficacy.1,13 Thus, a novel strategy is urgently needed to further improve the prognosis of these patients after failure to EGFR-TKIs.
Currently, immune-checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 interactions have shifted the therapeutical landscape of advanced/metastatic NSCLC without driver (e.g., EGFR, ALK, and ROS1) mutations. It has been incorporated into standard of care as a second-line treatment in advanced NSCLC.14,15,16,17 In addition, pembrolizumab or atezolizumab monotherapy is also approved as the first-line treatment in advanced NSCLC patients who have high PD-L1 expression in tumor tissues.18,19 However, anti-PD-(L)1 monotherapy did not show substantial survival benefits than standard chemotherapy in those who failed to EGFR-TKIs.20,21,22 Furthermore, clinical trials of osimertinib plus PD-1 blockade resulted in high rate of interstitial pneumonitis,23,24 suggesting that an alternative strategy is needed in this setting.
Comprehensive depiction of the tumor microenvironment (TME) might help clarify the mechanism of lower response and guide the subsequent therapeutic strategy. Previously, we and others have found that EGFR-mutant lung cancers are more likely to have lower PD-L1 expression, fewer CD8 + tumor-infiltrating lymphocytes (TILs), and lower tumor mutational burden (TMB) level than EGFR wild-type tumors.20,25 Additional evidence showed that chemotherapy-induced neoantigen release could modulate the TME to have a potentially synergistic effect with ICIs.26,27 Meanwhile, the addition of atezolizumab to bevacizumab plus chemotherapy showed superior efficacy in patients with EGFR-mutant NSCLC from IMPOWER 150 study,28 indicating potent clinical benefit of chemoimmunotherapy combination in this setting.
Toripalimab, a humanized IgG4κ mAb with hinge mutation (S228P) specific for human PD-1 receptor and blocks interactions of PD-1/PD-L1, has shown an acceptable safety profile and promising clinical activities in patients with advanced solid tumors in several phase-I/II studies29,30,31 and has been approved as the second-line setting for metastatic melanoma in 2018, China. Currently, several ongoing studies are investigating the combination of toripalimab with standard-of-care regimens as the first-line treatment in patients with NSCLC.
In order to evaluate the efficacy and safety of toripalimab plus carboplatin and pemetrexed in previously EGFR-TKI-treated patients with EGFR-mutant NSCLC, we conducted this open-label, single-arm phase-II trial and enrolled 40 patients from eight medical centers in China. We further performed integrated analysis of multi-omic data from PD-L1 expression, TMB, CD8 + TIL density, whole-exome, and transcriptome sequencing on tumor biopsies to identify the potential predictive biomarkers.
Results
Patient population
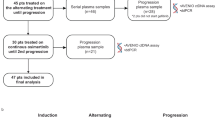
From April 2018 to March 2019, 40 patients were enrolled (Fig. 1). Baseline characteristics are summarized in Table 1. The median age was 58 years (range: 19–73), including 21 (52.5%) female and 19 (47.5%) male patients. Twenty-three (57.5%) patients had EGFR Exon 19 deletion (EGFR 19DEL) and 17 (42.5%) patients harbored Exon 21 L858R mutation (EGFR L858R). Twenty (50.0%) patients received gefitinib as first-line treatment, whereas 16 (40.0%) received icotinib and 4 (10.0%) received erlotinib. All the enrolled patients did not develop EGFR T790M mutation in the rebiopsy. Thirty patients received 360-mg dose of toripalimab treatment and 10 patients received the 240-mg dose.

CONSORT diagram for this phase-II study
Efficacy
As of October 22, 2020, the median follow-up time was 10.5 months. In total, 18 (45.0%) patients died, 22 (55.0%) discontinued treatment due to disease progression (PD), 3 (7.5%) discontinued due to adverse effects, and 2 (5.0%) patients remained on study. The overall confirmed objective-response rate (ORR) was 50.0% (95%CI 33.8–66.2), and the disease control rate (DCR) was 87.5% (95%CI 73.2–95.8). Tumor shrinkage was observed in 36 (90.0%) patients (Fig. 2a). The median duration of response (DOR) was 7.0 months (Fig. 2a), median PFS was 7.0 months (95%CI 4.8–8.4), and median overall survival (OS) was 23.5 (95% CI 18.0 to NR months) (Fig. 2c, d). Among patients who completed the induction treatment (n = 29), the ORR was 69.0%.

Summary of treatment response, progression-free and overall survival. a Maximal change of tumor size from baseline assessed by the investigator per RECIST v1.1 (n = 38). The length of the bar represents maximal decrease or minimal increase in target lesion(s). # Unconfirmed partial response classified as stable disease. * Patient with target lesion(s) reduction over 30% but with new lesion(s) or progression of nontarget lesion(s). b Change of individual tumor burden over time from baseline assessed by the investigator per RECIST v1.1 (n = 38). c Progression-free survival by RECIST v1.1 of all 40 enrolled patients. d Overall survival by RECIST v1.1 of all 40 enrolled patients. No. number
Subgroup analysis showed that the ORR was 58.8% in patients with EGFR L858R and 43.5% in patients with EGFR 19DEL (Supplementary Table S1). Patients with EGFR L858R had numerically longer median PFS (7.6 versus 5.4 months, P = 0.639) and OS (23.5 versus 18.0 months, P = 0.164) than patients with EGFR 19DEL (Supplementary Fig. S1a and S1b). Patients in 360 mg of toripalimab cohort (n = 30) and 240 mg of toripalimab cohort (n = 10) had similar ORR. Subgroup analysis of clinical characteristics, including age, gender, prior TKI treatment, and liver/central nervous system (CNS) metastasis, was seen in Supplementary Table S1. As to the subgroup analysis, we found that patients with LDH at normal range had numerically better ORR than those with LDH above the upper limit of normal (54.3% versus 20.0%, P = 0.34). In addition, three patients with liver metastasis had no objective response, whereas four out of six (66.7%) patients with CNS metastasis had partial response.
Safety
As data cutoff, 39 out of 40 (97.5%) of patients experienced treatment-related adverse events (TRAE, Table 2). The most common (≥20%) TRAE included 33 (82.5%) leukopenia, 28 (70.0%) neutropenia, 27 (67.5%) anemia, 21 (52.5%) elevated AST, 20 (50.0%) elevated ALT, 19 (47.5%) nausea, 19 (47.5%) thrombocytopenia, 15 (37.5%) decreased appetite, 11 (27.5%) constipation, and 10 (25.0%) asthenia. Grade 3 and above TRAE occurred in 26 patients (65.0%), including 16 (40.0%) neutropenia, 9 (22.5%) leukopenia, 4 (10%) thrombocytopenia, 2 (5%) of each anemia, pneumonia, and abnormal hepatic function, and 1 (2.5%) of each upper respiratory-tract infection, decreased appetite, maculo-papular rash, bone marrow suppression, febrile neutropenia, interstitial lung disease, and thoracic hemorrhage. Permanent treatment discontinuation of toripalimab due to TRAE occurred in 4 (10%) patients, while 15 (37.5%) patients had toripalimab delayed due to TRAE. The immune-related AEs (irAEs) included 16 (40.0%) thyroiditis, 12 (30.0%) skin toxicity, 10 (25.0%) hepatitis, 5 (12.5%) nausea, 3 (7.5%) uveitis, 1 (2.5%) pneumonitis, 1 (2.5%) conjunctivitis, 1 (2.5%) anemia, and 1 (2.5%) leukopenia. Grade 3 and above irAEs only occurred in 2 patients (5.0%), including 1 (2.5%) skin toxicity and 1 (2.5%) pneumonitis (Table 2).
Predictive role of PD-L1 expression, TMB, and CD8+TIL density
Tumor PD-L1 expression was tested in all 40 patients by using JS311 assay, which previously showed comparable results with 28-8, 22C3, and SP263 antibodies in NSCLC30. Patients of positive PD-L1 expression had numerically higher ORR (61.9% versus 36.8%, P = 0.204; Supplementary Table S1), longer median PFS (7.6 versus 5.8 months, P = 0.424; Supplementary Fig. S2a), and OS (NR versus 21.0, P = 0.052; Supplementary Fig. S2b) than negative PD-L1 expression. Among patients of PD-L1 expression ≥10%, the difference in ORR (75.0% versus 39.3%, P = 0.081) and PFS (median 8.1 versus 5.8 months, P = 0.697; Supplementary Fig. S2c) was more pronounced, while not in OS (median 21.1 versus 23.5 months, P = 0.424; Supplementary Fig. S2d). TMB was determined in 34 patients. Patients with high TMB (>median) had analogous ORR (57.1% versus 50.0%, P = 0.738), PFS (median 7.1 versus 5.1 months, P = 0.422; Supplementary Fig. S2e), and OS (median 23.5 versus 23.5 months, P = 0.541; Supplementary Fig. S2f) to those with low TMB (≤median). Intriguingly, patients with high CD8 + TIL density have numerically shorter PFS (median 5.8 versus 7.1 months, P = 0.726; Supplementary Fig. S2g) and OS (median 18.0 versus 27.1 months, P = 0.275; Supplementary Fig. S2h) than those with low CD8 + TIL density.
Whole-exome sequencing
Whole-exome sequencing (WES) was performed on both tumor biopsies and paired blood cells in 34 patients. WES identified 7048 genetic alternations, including 3505 missense mutations, 84 gene deletions, 123 rearrangements, 119 alternative splicing sites, 349 truncations, and 2748 gene amplifications. Having noticed the significant difference of gene alterations between patients with partial response (PR) versus non-PR (Fig. 3a), we inferred that genetic mutation might be utilized as predictive markers to this regimen. Prediction of genesets associated with efficacy was performed using Maftools::survGroup (v2.6.05) on the top 100 most frequently mutated genes.32 Considering the small size of data set, we set a relatively strict criteria (P ≤ 0.01) to screen genes or genes’ combination with predictive ability. We found that the prediction model will have an excellent effect when the number of components reaches two. In order to ensure the validity of our model, the minimum sample amount for each group should be more than 25% of the whole population (Fig. 3b).

Whole-exome sequencing. a Highlight mutated genes color-coded by the type of mutations in experimental samples. b According to the P value of PFS between groups, the top three combinations with different gene numbers were selected. The red line in the left panel shows the threshold of the filter. The white number on the barplot in the right panel represents the proportion of the corresponding sample in the total population. c Progression-free survival in patients with DSPP + TP53 comutation versus wild type (log-rank test). d Immune infiltration of DSPP + TP53 comutation versus wild type from EGFR-mutant samples obtained from TCGA (Wilcoxon signed-rank test). e Progression-free survival in patients with DSPP mutation versus wild type (log-rank test). f Immune infiltration of DSPP mutation versus wild type from EGFR mutant samples obtained from TCGA (Wilcoxon signed-rank test). PFS progression free survival; HR hazard ratio; *P < .05, **P < .01, ***P < .001, ****P < .0001
The double mutations of DSPP and TP53 (DT) were the most effective combination after screening, which were associated with significantly longer median PFS than wild type (9.3 versus 4.9 months, P = 0.008; Fig. 3c). Moreover, immune-infiltration analysis using CIBERSORTx in 354 EGFR-mutant patients from 18 projects of TCGA datasets showed that the DT double mutations have a significantly increased CD8+T cell and decreased M2 macrophage infiltration compared with wild-type tumors (Fig. 3d). Interestingly, DSPP could be used alone as a potential predictive biomarker (8.1 versus 5.3 months, P = 0.054; Fig. 3e), while TP53 alone could not (Supplementary Fig. S3a). Compared with TP53, similarity in the characteristics of immune infiltration between DSPP mutation and DT double mutations was observed (Fig. 3f), suggesting the potential predictive value of DSPP mutation rather than TP53 (Supplementary Fig. S3b). However, neither DT single nor double mutations cannot predict OS (Supplementary Fig. S3c–e).
Whole transcriptome sequencing
To discover the differential gene expression and characteristics of immune infiltration corresponding to different therapeutic effects, we conducted RNA-seq on 18 patients with adequate tissue samples. In general, the similarity between samples is relatively strong (Fig. 4a). Only 13 differentially expressed genes (DEGs) were found between two groups (P ≤ 0.05, log2fold change ≥2), including eight upregulated and five downregulated genes (Fig. 4b). Moreover, we found that the expression value of these 13 genes could not be used to distinguish two different therapeutic groups directly. Considering the small number of DEGs and analogous expression value between two groups, we tried to use support vector machines (SVM) to select candidate marker genes and then identify genes with high-accuracy classification performance (Supplementary Fig. S4a). The results showed that six components in a set could have the precise distinguishing ability (Supplementary Fig. S4b). The gene set possesses the best grouping ability that consisted of six genes, including RNF220, MAPK8IP2, PTGER2, P2RX4, ACADVL, and SPTSSB (Supplementary Fig. S4c). After 1000 times independent random grouping experiments on our data set, we found that five of the six test samples can be grouped correctly 985 times with the predictor mentioned above, and 951 of them can be grouped into all pairs. Notably, differential expression analysis between PR and non-PR groups at the network level by sample-specific networks (SSN) also validated the grouping ability of the six-gene set (Supplementary Fig. S4c).

Integrated analysis of whole exome and transcriptome sequencing. a Sample correlation heat map based on expression matrix; different colors represent different correlation coefficients. b Volcano plot of eight upregulated and five downregulated differentially expressed genes between PR and non-PR group, each of them labeled the names of the first five genes. c Immune analysis of PR versus non-PR group from our cohort (Wilcoxon signed-rank test). d M1/M2 macrophage ratio of PR versus non-PR from our cohort (Wilcoxon signed-rank test). e M1/M2 macrophage ratio of DSPP-mutant versus wild-type tumors from EGFR-mutant samples obtained from TCGA (Wilcoxon signed-rank test). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Then we focused on the analysis of distinct immune infiltration between PR and non-PR groups. We observed that patients with PR had significant lower CD8+T-cell abundance than those with non-PR (Fig. 4c). In addition, patients with PR had dramatically higher fractions of M1 macrophage than those with non-PR (Fig. 4c). Although there was no significant difference in the fraction of M2 macrophage between two groups, the ratio of M1/M2 was found to be markedly higher in PR group (P = 0.025; Fig. 4d), indicating the pivotal role of macrophage in determination of antitumor activity of PD-1 blockade plus chemotherapy in EGFR-mutant NSCLC.33
Integrated analysis of whole exome and transcriptome sequencing
Having noticed the potential predictive significance of DSPP mutation, we performed the integrated analysis of whole-exome and transcriptome-sequencing data from the TCGA database to explicate the biological mechanism of the association between DSPP mutation and therapeutic effect. First, we conducted differential expression analysis and pathway enrichment. In total, 1612 DEGs were identified (1536 upregulated and 76 downregulated; P ≤ 0.05, log2fold change ≥2, Supplementary Fig. S5a). After sorting the Enrichment Score (ES) estimated by GSEA data, top 20 upregulated and downregulated pathways were selected. We found that mitogen-activated protein kinase (MAPK) signaling pathway gene set was significantly downregulated (ES = -0.309) in the DSPP-mutant group (Supplementary Fig. S5b). Protein–protein-interaction network (score ≥ 900) analysis downloaded from STRING database showed that DSPP could establish the connection with MAPK1 and MAPK3 via the interaction of ITGAV, which are important regulatory genes in MAPK signaling pathway (Supplementary Fig. S5c). The relationship between these genes suggested that DSPP could affect the efficacy of immunotherapy through MAPK signaling pathway. Estimation of immune-infiltration features showed that DSPP-mutant tumors had remarkably higher M1/M2 ratio of macrophage (P < 0.001; Fig. 4e) than wild-type tumors. Moreover, DSPP mutation was associated with an increased IFN-γ signature gene expression in both our cohort (average score: 0.12 vs 0.08, P = 0.182; Supplementary Fig. S6a) and TCGA NSCLC cohort (average score: 0.49 vs 0.30, P < 0.001; Supplementary Fig. S6b).
Discussion
Currently, PD-1/PD-L1 blockade has become a backbone for the treatment in advanced NSCLC patients without driver mutations.34,35 Meanwhile, several studies attempted to evaluate the efficacy of PD-1/PD-L1 blockade monotherapy in NSCLC patients with EGFR mutations, either at the frontline setting in PD-L1 high-expression population or at later line setting after the failure of EGFR-TKI.36,37 All of them led to disappointing results. Combination of EGFR-TKIs and PD-1 blockade even resulted in high rate of interstitial pneumonitis and deemed not feasible for further clinical development.23,24 Moreover, anti-PD-1/PD-L1 monotherapy was also found to be associated with hyperprogressive disease in EGFR-mutant NSCLC.38 Therefore, an alternative strategy is urgently needed for patients with EGFR-mutant NSCLC who became refractory to EGFR-TKIs.
Previously, IMPRESS trial found that continuation of gefitinib plus chemotherapy did not show PFS benefit (5.4 versus 5.4 months) than chemotherapy alone in EGFR-mutant patients with advanced NSCLC and progressed from first-line EGFR-TKI.39 Thus, chemotherapy is the current standard of care in this setting with median PFS of 4–5 months and OS of 16–18 months. The present study is the first prospective study to evaluate the efficacy and safety of PD-1 blockade plus chemotherapy as second-line setting in patients with EGFR-mutant advanced NSCLC. We found that toripalimab plus carboplatin and pemetrexed had an ORR of 50% with a median PFS of 7.0 months, and a median OS of 23.5 months, which were superior than historical data of traditional chemotherapy. Consistently, subgroup analysis of patients with EGFR mutation from IMPOWER 150 study also demonstrated a superior efficacy of the addition of atezolizumab to bevacizumab and chemotherapy than the standard of care.40 Taken together, these findings suggest that the chemo-immunotherapy combination is a promising strategy in previously treated patients with EGFR-mutant advanced NSCLC.
The most common adverse events were leukopenia, neutropenia, anemia, elevated AST and ALT, nausea, and thrombocytopenia, which were consistent with the side effect of the similar chemo-immunotherapy combination in NSCLC patients without driver mutations.40 Interestingly, combination of EGFR-TKIs and PD-1 blockade would result in high rate of interstitial pneumonitis in patients with EGFR-mutant advanced NSCLC. However, we did not observe any unexpected AEs in this study. All of the immune-related AEs were consistent with previous studies on toripalimab in this study.29,30,31 These findings were reminiscent of the importance of the sequence of EGFR-TKI and immune-checkpoint inhibitors in this population. Notably, PD-1 blockade monotherapy-related thyroid toxicity was not prominent in this study, which also needs to be validated in a large-cohort study.
Biomarker analysis of PD-L1 expression, TMB level, and CD8 + TIL density showed that none of them could predict the efficacy of toripalimab plus carboplatin and pemetrexed. Patients with PD-L1 high expression had an enhanced ORR of 75.0%. Nevertheless, the ORR could reach to 36.8% in patients of negative PD-L1 expression. Similarly, we did not observe the association of TMB with response rate or survival. Notably, we found that patients with high CD8 + TIL density had an inferior median PFS and OS than those with low CD8 + TIL density, which was reiterated in the immune-infiltration analysis of whole-transcriptome sequencing data. Since tumor microenvironment contains not only CD8 + TILs but also immunosuppressive cells, these findings suggest that unraveling distinct tumor-immune microenvironment features of EGFR-mutant lung cancer41 might be helpful to identify the benefit population.
Subsequently, we performed an integrated analysis of whole-exome and transcriptome-sequencing data and found that patients with DSPP and TP53 comutations had significantly prolonged PFS than wild type. Among them, DSPP mutation, instead of TP53 mutation, was more potent to associate with superior PFS. Our finding that the DSPP mutation correlated with decreased M2 macrophage infiltration might partly be the possible explanation. As we know, M2 macrophage is immunosuppressive via suppressing the antitumor CD8 + T cells and secreting suppressive chemokine or cytokine. Additionally, our integrated analysis also found that MAPK signaling pathway was significantly downregulated in the DSPP-mutant group. Previous reports have shown that MAPK signaling pathway plays an important role in TCR signaling cascade42 and co-inhibition of MEK and PD-1/PD-L1 interaction is synergistic and resulted in a durable tumor regression.43 Therefore, its downregulation of MAPK signaling pathway might also attribute the superior efficacy in the patients with DSPP mutation, and the detailed mechanisms warrant investigation in the future.
We should mention that several limitations existed in this study. First, this is a single-arm phase II trial with a limited number of patients and all of them are Asian. Herein, these results should be cautiously interpreted. Second, only patients who failed from 1st/2nd generation of EGFR-TKIs and without T790M mutation were included. Therefore, the current findings could not be generalized to the population who had T790M mutation or received first-line osimertinib. Third, the primary endpoint was assessed by investigators in this study; therefore, the absence of central blinded review of ORR might have some bias. Last but not least, it has been reported that the PFS benefits with EGFR-TKI plus chemotherapy or anti-angiogenesis in the first-line setting. Therefore, these regimens might challenge the clinical relevance of the present study.
In summary, this phase-II study first provides evidence for the safety, efficacy, and potential predictive biomarkers of the combination of toripalimab with carboplatin and pemetrexed as second-line setting in patients with EGFR-mutant NSCLC, which will be further validated by the ongoing randomized phase-III trial (NCT03924050).
Patients and methods
Study design
This study was a multicenter, open-label, phase-II clinical trial evaluating the safety and clinical activity of toripalimab plus pemetrexed and carboplatin for the treatment of advanced NSCLC with EGFR-sensitive mutations after failure of EGFR-TKI therapy (NCT03513666). The study was approved by the institutional review board of participating centers and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Each subject provided written informed consent before any procedure of this study.
Patients’ eligibility
Eligible subjects should be aged 18–75 years old with histologically and/or cytologically confirmed advanced or recurrent NSCLC with EGFR-sensitive mutations, failed from prior first-line EGFR-TKI therapy (including gefitinib, erlotinib, icotinib, afatinib, or dacomitinib), did not harbor secondary EGFR T790M mutation, have at least one measurable lesion per response-evaluation criteria in solid tumors version 1.1 (RECIST v1.1), with eastern cooperative oncology group (ECOG) performance status of 0 or 1, and adequate organ and bone marrow function. Patients with previously treated asymptomatic brain metastasis were also allowed to enroll into this study. Exclusion criteria included mixed with small-cell lung-cancer component or squamous-cell carcinoma, other driver mutations with available targeting drugs, prior systemic chemotherapy, any active autoimmune disease or a history of autoimmune diseases, prior long-term systemic immunosuppressive therapy, or PD-1/PD-L1 blockade.
Treatment and endpoints
Two different doses (240 mg or 360 mg) of toripalimab were used for the initial safety and efficacy analysis in 10 patients and then chose one in the subsequent enrolled patients. Patients received toripalimab intravenously once every three weeks until PD, or intolerable side effects. During the induction phase, patients received up to six cycles of 500 mg/m2 pemetrexed plus AUC 5 carboplatin via intravenous infusion, once every three weeks. During the maintenance phase, patients received toripalimab plus 500 mg/m2 pemetrexed. The washout period between the end of EGFR-TKI and initiation of toripalimab was more than 28 days. Response was assessed every six weeks according to RECIST v1.1 locally by investigators. Patients who initially developed PD were allowed to stay on the study if the investigator considered that patients could benefit from the treatment. Treatment beyond the second PD was not allowed. Adverse events were assessed according to the national cancer institute common terminology criteria for adverse events, version 4.03, and monitored throughout and for 60 days after treatment discontinuation. The primary endpoint was ORR. The secondary end points included safety, DCR, PFS, and OS.
EGFR mutation detection
Tumor rebiopsy samples that have a conformed pathological diagnosis of NSCLC were collected for EGFR-mutation testing. EGFR (exons 18–22) was sequenced by using genomic DNA. Cycle sequencing of the purified polymerase chain reaction (PCR) products was carried out with PCR primers using the commercially available ADx Mutation Detection Kits (Amoy Diagnostics Company Ltd., Xiamen, China).
PD-L1 expression
Tumor rebiopsy sample was obtained from each patient before the initiation of this study to perform exploratory PD-L1 expression analysis. PD-L1 expression was detected by immunohistochemistry (IHC) staining with JS311 antibody using ventana benchmark autostainer. PD-L1-positive status was defined as the presence of membrane staining of any intensity in ≥1% of tumor cells. We adopted two different cutoffs of PD-L1 ≥ 1% or PD-L1 ≥ 10% to perform the biomarker analysis.
CD8+TIL density analysis
CD8+TIL density was assessed by using a mouse anti-CD8 monoclonal antibody (M7103, clone C8144B, DAKO). Lymphocytes with immunostained CD8 infiltrating within tumor region (central or marginal) were defined as CD8+TILs. On the basis of the percentage of CD8+TILs displayed within tumor region, we determined high/low CD8+TIL density (CD8+TIL+/−) with cutoff of 5%, which was analogous to the previous studies.44,45,46
WES and TMB analysis
WES was performed with SureSelect Human All Exon V6 kit (Agilent) on tumor biopsies and matched peripheral blood mononuclear cell (PBMC) samples. The detailed sequencing and analysis process were summarized in Supplemental Materials. Genomic alterations, including single-base substitution (SNV), short and long insertions/deletions (INDELs), copy number variants (CNV), and gene rearrangement and fusions, were assessed. The TMB was determined by analyzing somatic mutations, including coding-base substitution and INDELs per megabase (Mb).
Whole transcriptome sequencing
Total RNA was extracted from available tumor tissue samples and RNA quality and quantity were determined by capillary electrophoresis on Eukaryote Total RNA Pico chips (Agilent Technologies). The prepared libraries were sequenced on an Illumina HiSeq 2000 sequencer. The details bioinformatic analysis, including identification of DEGs, functional enrichment analyses, immune-infiltration estimation, SVM, and SSN, was summarized in Supplemental Materials.
Sample size determination and statistical analysis
This is a phase-II trial with a single-stage design. At one-sided significance level of 0.05, a total of 39 patients could provide 80% power to show the efficacy of toripalimab plus chemotherapy in the second-line setting with a target ORR of 50% versus 30% of standard chemotherapy using Clopper–Pearson method.
Safety and efficacy analyses included all patients who received ≥1 dose of study medication. ORR and its 95% exact CI were determined by Clopper and Pearson methodology. PFS and OS were plotted using the Kaplan–Meier method, with median and the corresponding two-sided 95% CIs reported. Duration of response was analyzed with Kaplan–Meier method with data from all responders. The data cutoff for analysis was October 22, 2020. Statistics analyses were performed with SAS or GraphPad Prism software.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Public data resources: The TCGA datasets, including COAD and READ, were downloaded from cBioPortal (http://www.cbioportal.org/).
References
Passaro, A., Jänne, P. A., Mok, T. & Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2, 377–391 (2021).
Yuan, M., Huang, L. L., Chen, J. H., Wu, J. & Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target Ther. 4, 61 (2019).
Zhou, C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 12, 735–742 (2011).
Mitsudomi, T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 11, 121–128 (2010).
Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246 (2012).
Mok, T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009).
Yang, J. C. et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 16, 141–151 (2015).
Wu, Y. L. et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 1454–1466 (2017).
Soria, J. C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Mok, T. S. et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 376, 629–640 (2017).
Soria, J. C. et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 16, 990–998 (2015).
Nishimura, T. et al. Second-line therapy with first- or second-generation tyrosine kinase inhibitors in EGFR-mutated non-small cell lung cancer patients with T790M-negative or unidentified mutation. Thorac. Cancer 12, 1067–1073 (2021).
Oxnard, G. R. et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 4, 1527–1534 (2018).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Brahmer, J. et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015).
Fehrenbacher, L. et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 387, 1837–1846 (2016).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
Herbst, R. S. et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N. Engl. J. Med. 383, 1328–1339 (2020).
Ng, T. L. et al. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non-small cell lung cancer. Cancer 125, 1038–1049 (2019).
Mazieres, J. et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann. Oncol. 30, 1321–1328 (2019).
Qiao, M. et al. Immune checkpoint inhibitors in EGFR-mutated non-small cell lung cancer: dusk or dawn? J. Thorac. Oncol 16, 1267–1288 (2021).
Oxnard, G. R. et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 31, 507–516 (2020).
Oshima, Y., Tanimoto, T., Yuji, K. & Tojo, A. EGFR-TKI-associated interstitial pneumonitis in nivolumab-treated patients with non-small cell lung cancer. JAMA Oncol. 4, 1112–1115 (2018).
Dong, Z. Y. et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology 6, e1356145 (2017).
Rottenberg, S., Disler, C. & Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 21, 37–50 (2021).
Petroni, G., Buque, A., Zitvogel, L., Kroemer, G. & Galluzzi, L. Immunomodulation by targeted anticancer agents. Cancer Cell 39, 310–345 (2021).
Reck, M. et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet. Respir. Med. 7, 387–401 (2019).
Wang, F. H. et al. Efficacy, safety, and correlative biomarkers of toripalimab in previously treated recurrent or metastatic nasopharyngeal carcinoma: a phase II clinical trial (POLARIS-02). J. Clin. Oncol. 39, 704–712 (2021).
Wang, Z. et al. Safety, antitumor activity, and pharmacokinetics of toripalimab, a programmed cell death 1 inhibitor, in patients with advanced non-small cell lung cancer: a phase 1 trial. JAMA Netw. Open 3, e2013770 (2020).
Yang, J. et al. Safety and clinical efficacy of toripalimab, a PD-1 mAb, in patients with advanced or recurrent malignancies in a phase I study. Eur. J. Cancer 130, 182–192 (2020).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Duan, Z. & Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target Ther. 6, 127 (2021).
Salas-Benito, D. et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discov., https://doi.org/10.1158/2159-8290.CD-20-1312 (2021).
Huang, M. Y., Jiang, X. M., Wang, B. L., Sun, Y. & Lu, J. J. Combination therapy with PD-1/PD-L1 blockade in non-small cell lung cancer: strategies and mechanisms. Pharm. Ther. 219, 107694 (2021).
Lisberg, A. et al. A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naive patients with advanced NSCLC. J. Thorac. Oncol. 13, 1138–1145 (2018).
Gainor, J. F. et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin. Cancer Res. 22, 4585–4593 (2016).
Fuentes-Antras, J., Provencio, M. & Diaz-Rubio, E. Hyperprogression as a distinct outcome after immunotherapy. Cancer Treat. Rev. 70, 16–21 (2018).
Mok, T. S. K. et al. Gefitinib plus chemotherapy versus chemotherapy in epidermal growth factor receptor mutation-positive non-small-cell lung cancer resistant to first-line gefitinib (IMPRESS): overall survival and biomarker analyses. J. Clin. Oncol. 35, 4027–4034 (2017).
Zhou, J. et al. An advanced non-small cell lung cancer patient with epidermal growth factor receptor sensitizing mutation responded to toripalimab in combination with chemotherapy after resistance to osimertinib: a case report. Transl. Lung Cancer Res. 9, 354–359 (2020).
van der Leun, A. M., Thommen, D. S. & Schumacher, T. N. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232 (2020).
D’Souza, W. N., Chang, C. F., Fischer, A. M., Li, M. & Hedrick, S. M. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol. 181, 7617–7629 (2008).
Ebert, P. J. R. et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621 (2016).
Tokito, T. et al. Predictive relevance of PD-L1 expression combined with CD8+ TIL density in stage III non-small cell lung cancer patients receiving concurrent chemoradiotherapy. Eur. J. Cancer 55, 7–14 (2016).
Yang, H. et al. Prognostic value of PD-L1 expression in combination with CD8(+) TILs density in patients with surgically resected non-small cell lung cancer. Cancer Med. 7, 32–45 (2018).
Jiang, T. et al. Genomic landscape and its correlations with tumor mutational burden, PD-L1 expression, and immune cells infiltration in Chinese lung squamous cell carcinoma. J. Hematol. Oncol. 12, 75 (2019).
Acknowledgements
This study is sponsored by Shanghai Junshi Biosciences. The authors thank the patients who participated in this study and their families. This work was supported by Junshi Medicine Co, Ltd. This study was also supported in part by grants from the National Natural Science Foundation of China (No. 81871865, 81874036, 81972167, 82102859, 31930022, 31771476, and 12026608), National Science and Technology Major Project (No. 2017YFA0505500), the Strategic Priority Project of Chinese Academy of Sciences (No. XDB38040400, XDB38020000), the Backbone Program of Shanghai Pulmonary Hospital (No. FKGG1802), Shanghai Pujiang Talent Plan (No. 2019PJD048), Shanghai Science and Technology Committee Foundation (NO. 19411950300), Shanghai Key disciplines of Respiratory (No. 2017ZZ02012), and the Shanghai Sailing Program (No. 20YF1407500).
Author information
Authors and Affiliations
Contributions
Conception and design: S.R., S.Y., and C.Z.. Provision of study materials or patients: S.R., J.Z., Y.Z., J.Z., Y.F., Y.S., X.L., H.Z., J.H., H.F., S.Y., and C.Z.. Collection and assembly of data: T.J., P.W., S.R., L.C., G.G., X.M., Z.B., Y.X., R.G., H.W., L.D., N.M., Y.Z., H.F., and S.Y.. Data analysis and interpretation: T.J., P.W., S.R., H.F., S.Y., J.W., L.C., and C.Z.. Paper writing: T.J., P.W., S.R., S.Y., J.W., L.C., and C.Z.. Final approval of the paper: All authors.
Corresponding authors
Ethics declarations
Competing interests
Honoraria to Dr. Caicun Zhou: Lilly, Roche, Boehringer Ingelheim, MSD, Hengrui Therapeutics, and QiLu Pharmaceutical; Consulting or advisory role: AmoyDx, Hengrui Therapeutics, Inovent, and QiLu Pharmaceutical. Other authors declared no potential conflicts of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, T., Wang, P., Zhang, J. et al. Toripalimab plus chemotherapy as second-line treatment in previously EGFR-TKI treated patients with EGFR-mutant-advanced NSCLC: a multicenter phase-II trial. Sig Transduct Target Ther 6, 355 (2021). https://doi.org/10.1038/s41392-021-00751-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00751-9
This article is cited by
-
Chemotherapy versus personalized therapy for EGFR mutant lung adenocarcinoma resistance to EGFR-tyrosine kinase inhibitors: a retrospective dual-center study
BMC Pulmonary Medicine (2024)
-
Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies
Signal Transduction and Targeted Therapy (2023)
-
Efficacy and safety of immune checkpoint inhibitors in post-TKI NSCLC patients harboring EGFR mutations
Journal of Cancer Research and Clinical Oncology (2023)
-
Liquid biopsy in lung cancer: significance in diagnostics, prediction, and treatment monitoring
Molecular Cancer (2022)
-
First-Line Treatment of Advanced Non-Small-Cell Lung Cancer with Immune-Checkpoint Inhibitors: New Combinations and Long-Term Data
BioDrugs (2022)
