
Abstract
Hypoxia-inducible factor-1 (HIF-1) plays essential roles in human diseases, though its central role in oxygen homoeostasis hinders the development of direct HIF-1-targeted pharmacological approaches. Here, we surveyed small-molecule compounds that efficiently inhibit the transcriptional activity of HIF-1 without affecting body homoeostasis. We focused on Mint3, which activates HIF-1 transcriptional activity in limited types of cells, such as cancer cells and macrophages, by suppressing the factor inhibiting HIF-1 (FIH-1). We identified naphthofluorescein, which inhibited the Mint3–FIH-1 interaction in vitro and suppressed Mint3-dependent HIF-1 activity and glycolysis in cancer cells and macrophages without evidence of cytotoxicity in vitro. In vivo naphthofluorescein administration suppressed tumour growth and metastasis without adverse effects, similar to the genetic depletion of Mint3. Naphthofluorescein attenuated inflammatory cytokine production and endotoxic shock in mice. Thus, Mint3 inhibitors may present a new targeted therapeutic option for cancer and inflammatory diseases by avoiding severe adverse effects.
Similar content being viewed by others

Introduction
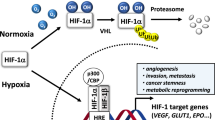
Oxygen is essential for eukaryotes, and cellular oxygen-sensing mechanisms play important roles in maintaining homoeostasis and in developing various diseases, such as cancer and inflammatory diseases. Among the cellular oxygen-sensing mechanisms, hypoxia-inducible factor-1 (HIF-1) is one of the most important transcriptional factors that promotes the expression of various genes related to metabolic pathways in glycolysis, angiogenesis, erythropoiesis, among others, in adaptive responses to hypoxic conditions1,2,3,4,5. HIF-1 is a heterodimeric transcription factor that comprises a regulatory α-subunit and a constitutive β-subunit. HIF-1α is negatively regulated by two types of hydroxylases, a HIF prolyl hydroxylase domain-containing protein 1−3 (PHD1−3) and the factor inhibiting HIF-1 (FIH-1), in an oxygen-dependent manner. PHD1−3 hydroxylates two proline residues of HIF-1α and, thereby, promotes proteasomal degradation of the HIF-1α protein. In turn, FIH-1 hydroxylates an asparagine residue of HIF-1α and, thereby, inactivates the transcriptional activity of HIF-1α1,3,5. Many studies have demonstrated that HIF-1 inhibition is effective for cancer therapy in various experimental models6; however, a specific HIF-1 inhibitor is not clinically available. HIF-1 is essential not only for cancer and inflammatory diseases but also for body homoeostasis; HIF-1α knockout in mice is embryonically lethal at day E11 due to cardiovascular malfunctions and neural tube defects, whereas HIF-1α conditional knockout mice show various defects in this regard7,8,9,10,11,12. These results indicate that HIF-1 inhibition itself can cause many adverse effects within the body. In particular, systemic HIF-1 inhibition would be contraindicated for individuals with heart failure because HIF-1 plays a protective role in cardiovascular diseases13. Therefore, drugs that target disease-specific regulatory mechanisms of HIF-1 activation are required.
Munc18-1-interacting protein 3 (Mint3) activates HIF-1 even under normoxic conditions by binding to FIH-1 and, thereby, interfering with the interaction between FIH-1 and HIF-1α14,15. Mint3 has a unique naturally disordered N-terminal region and a C-terminal region with one phosphotyrosine-binding and two PDZ domains conserved among other Mint family proteins, and the N-terminal region of Mint3 binds to FIH-114,16,17. Mint3 requires the expression of a transmembrane metalloproteinase, MT1-MMP, to bind to FIH-115,18,19; moreover, the mTORC1 signalling pathway and the monooxygenase NECAB3 promote the interaction between Mint3 and FIH-1 in cells from some types of cancer20,21. Mint3 is expressed ubiquitously, whereas MT1-MMP is highly expressed in cancer cells and in activated macrophages15. Thus, Mint3-mediated HIF-1 activation occurs only in these cells. Furthermore, the Km values of FIH-1 and PHDs for O2 are about 90 and 230 μM, respectively, in vitro22, indicating that Mint3-dependent HIF-1 activation can be observed under normoxic and moderate hypoxic conditions where FIH-1 can hydroxylate HIF-1α. Therefore, Mint3 is a unique molecule that activates HIF-1 in specific cell types within limited microenvironments. Reflecting these properties, Mint3 knockout (KO) mice show no apparent abnormality compared with HIF-1α knockout mice23,24. However, Mint3 KO mice showed resistance against acute inflammatory diseases, such as lipopolysaccharide (LPS)-induced endotoxic shock and influenza-induced acute pneumonia, and suppression of cancer metastasis due to attenuated hyperactivation of macrophage-lineage cells23,25,26,27. In cancer cells, Mint3 depletion suppresses tumour growth of various types of cancer, such as breast cancer, pancreatic cancer, lung cancer, and fibrosarcoma, and enhances chemosensitivity in cancer cells18,20,21,28. These results indicate that Mint3 inhibitors may present a new targeted therapeutic option for cancer and inflammatory diseases by avoiding severe adverse effects.
In this study, to develop Mint3 inhibitors, we first screened compounds that can bind to the N-terminal region of Mint3 using the chemical arrays of 23,275 compounds.
Results
Screening for small-molecule compounds that inhibit Mint3
We initially surveyed compounds that bind to the N-terminus of Mint3 using a chemical array of 23,275 compounds (Fig. 1a) and identified one hit compound (Fig. 1b, compound #1). By using the HIF-1 reporter assay system (Fig. 1a) and HT1080 cells, where Mint3 activates HIF-1 even during normoxia, we evaluated whether compound #1 can suppress HIF-1 transcriptional activity21. Compound #1 showed an approximately 40% reduction in HIF-1 transcriptional activity at a concentration of 10 μM in HT1080 cells (Fig. 1c). To further explore potent Mint3 inhibitors, 18 compounds that were structurally similar to compound #1 were collected and subjected to the HIF-1 reporter assay in HT1080 cells (Supplementary Table 1). Compound #9 was excluded from further evaluation because it showed severe toxicity to HT1080 cells, and we observed, by using a microscope, that almost all the cells treated with compound #9 were detached from the plate. Compounds #5, #8, #11, and #19 significantly suppressed HIF-1 transcriptional activity (Fig. 1d). Among these compounds, compound #19, naphthofluorescein (Fig. 1e), showed the most profound suppression of HIF-1 transcriptional activity. Serial dilution of naphthofluorescein revealed that naphthofluorescein suppressed the HIF-1 reporter activity in a concentration-dependent manner (Fig. 1f). Therefore, we further assessed the biological actions of this compound.

a Schematic illustration of the screening process for Mint3 inhibitors. Mint3-binding compounds were identified using chemical arrays in the first screening. Then, using the luciferase assay in the second screening, hit compounds were tested to determine whether they inhibited HIF-1 transcriptional activity. b Structure of the hit compound, NPD7801 (compound #1). c Luciferase assay of HIF-1 activity in HT1080 cells treated with compound #1 at the indicated concentrations. d Luciferase assay of HIF-1 activity in HT1080 cells treated with 10 μM compound #1 and the analogues of compound #1 (#2–19). $ indicates no detection of luciferase activity due to cell death. e Structure of NPD8369 (compound #19), naphthofluorescein (Naph). f Luciferase assay for HIF-1 activity in HT1080 cells treated with DMSO or Naph at the indicated concentrations for 24 h. In (c), (d), and (f), error bars indicate SD (n = 3). Data were analysed using Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
Naphthofluorescein disrupts Mint3–FIH-1 interaction in vitro and attenuates HIF-1 activity in cancer cells
Direct interaction of Mint3 with FIH-1 activates HIF-1 function14,18. We used a pull-down assay to ascertain whether naphthofluorescein can block the interaction between Mint3 and FIH-1. Naphthofluorescein completely abolished the interaction between GST-FIH-1 and His6-Mint3 in vitro (Fig. 2a). Next, the specificity of naphthofluorescein for Mint3 was examined by the HIF-1 reporter assay using control and Mint3 knockdown HT1080 cells (Fig. 2b). Naphthofluorescein reduced HIF-1 reporter activity by approximately 60% in control siRNA (siGFP)-transfected HT1080 cells (Fig. 2c, siGFP). In turn, Mint3 knockdown HT1080 cells showed reduced HIF-1 reporter activity, and naphthofluorescein did not further suppress the HIF-1 reporter activity in these cells (Fig. 2c, siMint3#1, #2). Overexpression of Mint3 and knockdown of FIH-1 also cancelled the suppression of HIF-1 reporter activity by naphthofluorescein (Fig. 2d‒g). These results suggest that naphthofluorescein decreases HIF-1 reporter activity in a Mint3–FIH-1 axis-dependent manner.

a Pull-down assay of GST-FIH-1 and His6-Mint3 in the presence of DMSO or naphthofluorescein (Naph; 10 μM). b Mint3 expression in HT1080 cells transfected with siRNAs against green fluorescent protein (GFP; siGFP) and Mint3 (siMint3#1, #2). c Luciferase assay for HIF-1 activity in siRNA-transfected HT1080 cells treated with DMSO or Naph (10 μM). d V5-tagged Mint3 expression in HT1080 cells transfected with mock or V5-tagged Mint3 expression vector. e Luciferase assay for HIF-1 activity in mock or V5-tagged-Mint3 expressing HT1080 cells treated with DMSO or Naph (10 μM). f Mint3 expression in HT1080 cells transfected with siRNAs against GFP (siGFP) and FIH-1 (siFIH-1#1, #2). g Luciferase assay for HIF-1 activity in siRNA-transfected HT1080 cells treated with DMSO or Naph (10 μM). h–n mRNA levels of glycolysis-related HIF-1 target genes (h–l) and HIF1A (m), and 18S rRNA levels (n) in HT1080 cells treated with DMSO or Naph at the indicated concentration for 24 h. o Cell growth assay of HT1080 cells treated with DMSO or Naph at the indicated concentrations. In (c), (e), (g–o), error bars indicate SD (n = 3). Data were analysed using Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. NS not significant.
Naphthofluorescein has been reported to inhibit the conversion of MT1-MMP from a pro-form to a mature form by furin, with an IC50 of 12 μM 29. Thus, we examined whether furin inhibition by naphthofluorescein affects HIF-1 activity in HT1080 cells. At a concentration of 10 μM, naphthofluorescein did not increase the pro-form of MT1-MMP in HT1080 cells, whereas furin inhibitors did not affect HIF-1 activity at a concentration that increased the pro-form of MT1-MMP in HT1080 cells (Furin-i I at 20 μM and Furin-i II at 10 μM) (Supplementary Fig. 1a, b). Mint3 has also been reported to control the cellular localisation of overexpressed furin30. However, neither Mint3 knockdown nor naphthofluorescein treatment affected endogenous furin localisation at the Golgi apparatus in HT1080 cells (Supplementary Fig. 1c, d). Thus, naphthofluorescein suppressed HIF-1 activity in HT1080 cells independent of furin inhibition under our experimental conditions. FIH-1 has been reported to hydroxylate another HIFα subunit, HIF-2α, much less effectively compared with HIF-1α due to differences in amino acid sequences around the target asparagine residue22,31. We also confirmed that overexpression of FIH-1 suppressed HIF-2α reporter activity less effectively than HIF-1α in HT1080 cells (Supplementary Fig. 1e, f). Consistent with these results, naphthofluorescein treatment also showed little or no suppression of HIF-2α reporter activity in HT1080 cells (Supplementary Fig. 1g). Thus, naphthofluorescein shows selectivity for HIF-1 rather than for HIF-2.
Next, we examined whether naphthofluorescein affected the expression of HIF-1 target genes in HT1080 cells. Consistent with our observations in the HIF-1 reporter assay, naphthofluorescein suppressed the expression of HIF-1-target glycolysis-related genes in HT1080 cells in a concentration-dependent manner, whereas HIF-1α mRNA and 18S rRNA expressions were unaffected (Fig. 2h–n). FIH-1 knockdown also attenuated the suppression of HIF-1 target gene expression by naphthofluorescein in HT1080 cells (Supplementary Fig. 1h–m), indicating that naphthofluorescein suppressed HIF-1 target gene expression via the Mint3–FIH-1 axis. Mint3 depletion attenuates the in vivo growth of HT1080 cells but does not affect cell proliferation in vitro20,21. Likewise, at a concentration of 10 μM, naphthofluorescein did not affect the proliferation of HT1080 cells but suppressed HIF-1 reporter activity and the expression of endogenous glycolysis-related genes (Fig. 2o). Taken together, naphthofluorescein inhibits the Mint3-dependent transcriptional activity of HIF-1 without causing non-specific cell toxicity in HT1080 cells.
Naphthofluorescein attenuates Mint3-mediated glycolysis in MDA-MB-231 cells and macrophages
In addition to HT1080 cells, Mint3 promotes glycolysis in breast cancer MDA-MB-231 cells and in murine macrophages18,23,32. Thus, by measuring the levels of lactate, an end product of glycolysis, we examined whether naphthofluorescein can suppress glycolysis in these cells. Mint3 knockdown (siMint3#1, #2) MDA-MB-231 cells showed reduced lactate production compared to control (siGFP) cells (Fig. 3a, b), as previously reported18. Naphthofluorescein decreased lactate production in control MDA-MB-231 cells to the levels in Mint3 knockdown cells but did not further affect the lactate levels of Mint3 knockdown cells (Fig. 3b). Overexpression of exogenous Mint3 cancelled the decreased lactate production by naphthofluorescein in MDA-MB-231 cells (Fig. 3c, d). Similar to MDA-MB-231 cells, that naphthofluorescein decreased lactate production in WT macrophages to the levels in Mint3 KO cells, but did not further affect the lactate levels of Mint3 KO cells (Fig. 3e, f), show the reduced ATP levels due to defects of glycolysis23,26,32. Furthermore, naphthofluorescein reduced ATP levels in WT macrophages but not in Mint3 KO cells (Fig. 3g). Thus, naphthofluorescein suppresses Mint3-dependent glycolysis in MDA-MB-231 cells and in macrophages.

a Mint3 expression in MDA-MB-231 cells transfected with siRNAs against green fluorescent protein (GFP; siGFP) and Mint3 (siMint3#1, #2). b Lactate production of control and Mint3 knockdown MDA-MB-231 cells treated with DMSO or naphthofluorescein (Naph; 10 μM). c V5-tagged Mint3 expression in MDA-MB-231 cells transfected with mock or V5-tagged Mint3 expression vector. d Lactate production of mock or V5-tagged Mint3 expressing MDA-MB-231 cells treated with DMSO or Naph (10 μM). e Mint3 expression in wild-type (WT) and Mint3 knockout (KO) bone-marrow-derived macrophages. f Lactate production of WT and Mint3 KO macrophages treated with DMSO or Naph (10 μM). g ATP levels of WT and Mint3 KO macrophages treated with DMSO or Naph (10 μM). In (b), (d), (f), and (g), error bars indicate SD (n = 3). Data were analysed using Student’s t test. *p < 0.05, **p < 0.01. NS not significant.
Administration of naphthofluorescein attenuates the interaction between Mint3 and FIH-1 and the expression of HIF-1 target genes in vivo
To understand the in vivo effects of naphthofluorescein, we first evaluated the acute toxicity of naphthofluorescein in mice. The experiments were conducted in accordance with the institutional ethical guidelines for animal experiments. Naphthofluorescein 100 mg/kg body weight (b.w.) was intraperitoneally injected into C57BL/6 mice for 2 weeks (5-day/2-day injection-rest cycle per week). The mice receiving naphthofluorescein did not show weight loss (Fig. 4a) or apparent histological abnormalities in the lung, liver, and kidney (Supplementary Fig. 2a). Blood biochemical analyses did not show any significant difference between vehicle- and naphthofluorescein-treated mice (Supplementary Table 2). Thus, the administration of naphthofluorescein does not cause severe adverse effects for at least 2 weeks in mice.

a Body weight of mice with vehicle or naphthofluorescein (Naph; 100 mg/kg b.w.) injection (n = 6 per group). b Administration of Naph attenuates the interaction between Mint3 and FIH-1 in tumours. Tumours of HT1080 cells expressing FLAG-tagged FIH-1 were collected from mice injected vehicle or Naph (n = 5 per group) 3 h after the injection. FLAG-tagged FIH-1 was immunoprecipitated (IP FLAG) from tumour lysates, and precipitates were analysed by western blotting using the indicated antibodies. c–i Expression of HIF-1 target genes (c–g), HIF1A (h), and 18S rRNA (i) in tumours of HT1080 cells 6 h after Naph injection. Error bars indicate the SD (n = 10). Data were analysed using the Mann–Whitney U test.
Next, we addressed the question of whether naphthofluorescein can block the interaction between Mint3 and FIH-1 even in vivo. Nude mice with tumours of HT1080 cells expressing FLAG-tagged FIH-1 were injected with vehicle or naphthofluorescein, FLAG-tagged FIH-1 was immunoprecipitated from tumour lysates 3 h after the injection, and co-precipitated Mint3 was detected by western blotting. Administration of naphthofluorescein indeed attenuated the interaction between FLAG-tagged FIH-1 and Mint3 even in vivo (Fig. 4b). We also confirmed that the administration of naphthofluorescein did not affect the conversion of the potential furin substrates MT1-MMP and integrin α5 33,34 from their pro-forms to the mature forms in these tumours (Supplementary Fig. 2b).
Subsequently, we examined whether naphthofluorescein can suppress the expression of HIF-1 target genes in tumours. Nude mice with tumours of HT1080 cells were injected with vehicle or naphthofluorescein, and gene expression in tumours was analysed 8 h after the injection. The expression of the tested HIF-1 target genes was apart from PGK1 significantly reduced in tumours from naphthofluorescein-injected mice compared to that in tumours from vehicle-injected mice, and HIF-1α mRNA levels were not affected in these tumours (Fig. 4c–i). In turn, naphthofluorescein administration could not decrease the expression of these genes in tumours of Mint3-overexpressing HT1080 cells (Supplementary Fig. 2c–g). These results indicate that naphthofluorescein suppresses the expression of HIF-1-target glycolysis-related genes by targeting Mint3 in vivo.
Naphthofluorescein attenuates tumour growth and augments the effect of anticancer drugs in vivo
Mint3 knockdown attenuates the tumour growth of various types of cancers, such as breast cancer, pancreatic cancer, lung cancer, and fibrosarcoma, in mice18,20,21,28. Thus, we first examined whether naphthofluorescein suppresses the tumour growth of murine triple-negative breast cancer E0771 cells in immunocompetent C57BL/6 mice. Naphthofluorescein administration for 5 days a week strikingly suppressed the tumour growth of subcutaneously injected E0771 cells (Fig. 5a). By contrast, in immunodeficient nude mice, suppression of E0771 tumour growth by naphthofluorescein was moderate compared to that observed in immunocompetent mice (Supplementary Fig. 3), implying T-cell-dependent and -independent anti-tumour effects of naphthofluorescein. Further assessment of the T-cell-dependent anti-tumour effects of naphthofluorescein showed that more CD8+ T cells infiltrated the tumours of naphthofluorescein-administered immunocompetent mice compared with tumours of vehicle-administered mice (Fig. 5b).

a Tumour growth of E0771 cells in C57BL/6 mice (n = 8) treated with vehicle or naphthofluorescein (Naph; 100 mg/kg b.w.). Mice were intraperitoneally injected with vehicle or Naph five times a week (top). Photos of tumours at day 14 after initiation of therapy. Scale bar = 1 cm (bottom); tumour volumes of E0771 cells in mice. b Immunohistochemical (IHC) analysis of CD8α in tumours of E0771 cells treated with vehicle or Naph. Scale bars = 100 μm. c, d Tumour growth of MDA-MB-231 (c) and AsPC-1 (d) cells in immunodeficient mice treated with vehicle or Naph (100 mg/kg b.w.). Mice were intraperitoneally injected with vehicle or Naph five times a week (n = 6). e, f Tumour growth of MDA-MB-231 (e) and AsPC-1 (f) cells in immunodeficient mice treated with vehicle or Naph (100 mg/kg b.w.) in combination with vehicle, doxorubicin (DOX; 2 mg/kg b.w.), or gemcitabine (GEM; 20 mg/kg b.w.) twice a week (n = 5). g, h Immunostaining of cleaved caspase-3 in tumours of MDA-MB-231 from vehicle- or Naph-treated mice in combination with DOX at day 14 after initiation of therapy. g Representative photos. h Cleaved caspase-3-positive areas were counted in tumour sections (n = 18, from six tumours per group). In (a), (c–f), and (h), error bars indicate the SEM. Data were analysed using the Mann–Whitney U test. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, naphthofluorescein was tested in xenograft models of human triple-negative breast cancer MDA-MB-231 and pancreatic cancer AsPC-1 cells. The administration of naphthofluorescein for 5 consecutive days per week significantly attenuated tumour growth of MDA-MB-231 and AsPC-1 cells in immunodeficient mice (Fig. 5c, d). In turn, naphthofluorescein did not attenuate tumour growth of FIH-1-depleted MDA-MB-231 cells (Supplementary Fig. 3b–d). Thus, naphthofluorescein can suppress tumour growth in human cancer cells in an FIH-1-dependent manner. HIF-1 is involved in chemoresistance, and combinations of HIF-1 inhibitors and chemotherapy show strong anti-tumour effects4,6. In addition, Mint3 depletion suppresses chemoresistance in pancreatic cancer cells28. Therefore, we examined whether the combination of naphthofluorescein and chemotherapy showed additive/synergistic effects on tumour suppression. Naphthofluorescein was injected into tumour-bearing mice with or without cytotoxic chemotherapeutic reagents (doxorubicin for MDA-MB-231 and gemcitabine for AsPC-1 cells) twice a week. In these treatment regimens, naphthofluorescein or chemotherapeutic agents alone did not effectively suppress tumour growth. However, the combination of naphthofluorescein with doxorubicin or gemcitabine strikingly suppressed the tumour growth of MDA-MB-231 and AsPC-1 cells, respectively (Fig. 5e, f). We confirmed that the combined administration of naphthofluorescein and doxorubicin remarkably increased the apoptotic marker cleaved caspase-3-positive area in tumours of MDA-MB-231 cells (Fig. 5g, h). Taken together, naphthofluorescein improves the efficacy of chemotherapy in xenograft models.
Naphthofluorescein attenuates host Mint3-mediated lung metastasis
Metastasis involves not only cancer cells but also host cells such as immune and endothelial cells35,36,37,38. Mint3 in host cells, especially in inflammatory monocytes, promotes metastatic niche formation in the lung25,26. Thus, using experimental lung metastasis models of mouse melanoma B16F10 cells, we examined whether naphthofluorescein inhibits host Mint3-dependent metastasis (Fig. 6a). Naphthofluorescein significantly suppressed lung metastasis of B16F10 cells compared to vehicle (Fig. 6b, c) in a dose-dependent manner (Fig. 6d). Similarly, naphthofluorescein suppressed lung metastasis of mouse lung cancer Lewis lung carcinoma (LLC) cells (Supplementary Fig. 4a, b). Importantly, naphthofluorescein did not further suppress the lung metastasis of B16F10 cells in Mint3 KO mice (Fig. 6e, f), indicating that the effect of naphthofluorescein on metastasis is attributable to host Mint3, which promotes metastatic niche formation in the lung by recruiting inflammatory monocytes and inducing the expression of E-selectin in endothelial cells that are adjacent to cancer cells in metastasis-affected lungs26. Thus, we examined whether naphthofluorescein suppresses this metastatic niche formation. Naphthofluorescein suppressed both recruitment of inflammatory monocytes and E-selectin expression in metastatic lung cancer (Fig. 6g, h). Mint3 depletion attenuates chemotaxis toward CCL2 in macrophages/inflammatory monocytes due to the defect in ATP production via glycolysis without affecting the expression levels of CCR2 in macrophages, those of CCL2 in metastatic lungs, and serum in tumour-inoculated mice23,26. Similarly, naphthofluorescein did not affect the expression levels of CCR2 in macrophages, those of CCL2 in metastatic lungs, and serum of tumour-inoculated mice (Supplementary Fig. 4c–e). Thus, naphthofluorescein administration phenocopies the host Mint3 depletion in lung metastatic niche formation.

a Schematic illustration of the experimental lung metastasis assay of B16F10 cells. b Photos of metastatic lungs from vehicle- or naphthofluorescein-treated (Naph; 100 mg/kg b.w.) mice. Scale bars = 1 cm. c Number of metastatic foci of B16F10 cells in the lungs from mice treated with vehicle or Naph at the indicated amount (n = 6 mice per group). d Number of metastatic foci of B16F10 cells in the lungs from mice treated with Naph at the indicated dose (n = 5–6 mice per group). e Photos of metastatic lungs from vehicle- or Naph-treated WT and Mint3 KO mice. Scale bar = 1 cm. f Number of metastatic foci of B16F10 cells in the lungs from vehicle- or Naph-treated (100 mg/kg b.w.) WT and Mint3 KO mice (n = 6 mice per group). g Flow cytometric analysis of inflammatory monocytes (IMs) in the lungs at 4 h after intravenous injection of B16F10 cells into vehicle- or Naph-treated (100 mg/kg b.w.) mice (n = 6 per group). h Immunostaining analysis of E-selectin (red) expression in the lungs of vehicle- or Naph-treated mice 6 h after intravenous injection with fluorescein-labelled B16F10 cells (green). Nuclei are stained with Hoechst 33342 (blue). (Left) Representative photos of lungs from vehicle-treated mice. (Right) Quantitative analysis of E-selectin-positive cells in the lungs (n = 9, from three tumours per group). In (c), (d), and (f−h), data are presented as the mean ± SEM. Data were analysed using the Mann–Whitney U test. **p < 0.01. NS not significant.
Naphthofluorescein attenuates endotoxic shock and inflammatory cytokine production in macrophages
Naphthofluorescein inhibited host Mint3 in experimental metastasis models. This prompted us to evaluate the effects of naphthofluorescein in other Mint3-related disease models. Mint3 KO mice are resistant to LPS-induced endotoxic shock due to decreased macrophage hyperactivation23. Thus, we examined whether the administration of naphthofluorescein attenuates LPS-induced endotoxic shock. Pretreatment with naphthofluorescein significantly improved survival of LPS-injected WT mice, similar to that of mice with genetic depletion of Mint3 (Fig. 7a, b). Furthermore, naphthofluorescein pretreatment decreased serum levels of inflammatory cytokines, such as tumour necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-12p70, after LPS injection in WT mice (Fig. 7c–e), similar to that in the Mint3 KO mice phenotype23. Supporting these in vivo observations, naphthofluorescein decreased TNF-α and IL-6 production in LPS-stimulated WT macrophages to levels observed in Mint3 KO macrophages but did not further decrease cytokine production in LPS-stimulated Mint3 KO macrophages (Fig. 7f, g). Thus, naphthofluorescein specifically inhibits Mint3-dependent inflammatory cytokine production in macrophages, and naphthofluorescein pretreatment attenuates LPS-induced cytokine storm and improves the survival of mice with LPS-induced endotoxic shock.

a Schematic illustration of LPS-induced endotoxic shock experiments. b Survival of LPS-injected mice treated with vehicle or naphthofluorescein (Naph; 100 mg/kg b.w.; n = 9 per group). Data were analysed using the log-rank test. *p < 0.05. NS not significant. c–e Serum TNF-α (c), IL-6 (d), and IL-12p70 (e) levels from LPS-injected mice treated with vehicle or Naph. Data are presented as the mean ± SEM (n = 6). Data were analysed using the Mann–Whitney U test. *p < 0.05, **p < 0.01. f, g TNF-α (f) and IL-6 (g) production in LPS-stimulated WT and Mint3 KO macrophages with or without Naph (10 μM). Data are presented as the mean ± SD (n = 3). Data were analysed using Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. NS not significant. h Schematic diagram illustrating how Naph attenuates tumour growth, metastasis, and endotoxic shock. Naph disrupts the interaction between Mint3 and FIH-1, and liberated FIH-1 inhibits HIF-1 activity in cancer cells and macrophages, resulting in attenuation of tumour growth, metastasis, and endotoxic shock in vivo. Direct and indirect furin suppression by Naph might also contribute to these in vivo effects.
Discussion
We surveyed the range of small-molecule compounds that inhibit Mint3-mediated HIF-1 activation and identified naphthofluorescein, which blocked the interaction between Mint3 and FIH-1 in vitro and suppressed Mint3-dependent HIF-1 activity and glycolysis in cancer cells and macrophages without causing cytotoxicity in vitro. In vivo, naphthofluorescein decreased the expression of HIF-1 target genes in tumours without apparent adverse effects. Furthermore, naphthofluorescein administration suppressed tumour growth, metastasis, and LPS-induced endotoxic shock in mice. Especially, naphthofluorescein did not show any effects in Mint3 KO mice on metastasis and endotoxic shock models and on HIF-1 target gene expression in tumours of Mint3-overexpressing HT1080 cells, indicating the specificity of naphthofluorescein for Mint3 in vivo (Fig. 7h). Mint3 enhances the transcriptional activity of HIF-1α by suppressing the HIF-1 inhibitor FIH-1 14,15. Although their pathophysiological roles are not well understood, FIH-1 can hydroxylate not only HIF-1α but also other proteins with ankyrin repeat domains, such as Notch and IκBα39,40,41,42. Thus, Mint3 might suppress the hydroxylation of these abovementioned proteins by FIH-1; however, we cannot exclude the possibility that FIH-1 target proteins other than HIF-1α partially contribute to the anti-tumour and anti-inflammatory effects of naphthofluorescein. Naphthofluorescein suppressed tumour growth of MDA-MB-231 in an FIH-1 dependent manner. In turn, naphthofluorescein suppressed host Mint3-mediated metastasis and LPS-induced endotoxic shock in vivo, but we could not completely exclude in this study the possibility that naphthofluorescein might suppress in vivo Mint3-mediated metastasis and endotoxic shock independently from FIH-1. Further studies using FIH-1 knockout or conditional knockout mice may clarify the specificity of naphthofluorescein effects on FIH-1 in metastasis and endotoxic shock models. Naphthofluorescein and Mint3 have been reported to control furin activity/localisation, though we could not reproduce these effects in our experimental conditions29,30. Furin is involved in various biological events by targeting various substrates including MT1-MMP and integrin α5, and furin knockout in mice is embryonically lethal33,34,43. In turn, Mint3 knockout mice show no apparent abnormality23,24,44. Thus, Mint3 is not likely an essential regulator of furin, at least not during embryogenesis. However, we do not exclude the possibility that furin inhibition partially contributed to the anti-tumour effects of naphthofluorescein in Mint3-dependent and -independent manners (Fig. 7h).
Intriguingly, naphthofluorescein suppressed the tumour growth of E0771 cells in an immunity-dependent manner (Fig. 5a, b). The mechanism whereby Mint3 contributes to anti-tumour immunity remains unclear; however, Mint3 controls the inflammatory function of monocytes and, thereby, promotes metastasis25,26. Thus, Mint3 in macrophage-lineage cells might contribute to anti-tumour immunity. In tumour tissues, tumour-associated macrophages (TAMs) polarise to the M2-like subtype which suppresses anti-tumour immunity45. HIF-1α promotes M2-like polarisation in TAMs46, implying that Mint3 might also control M2-like polarisation in TAMs through HIF-1 activation, which we are investigating currently. In contrast, Mint3 depletion in cancer cells attenuates tumour growth18,20,21,28. Thus, the anti-tumour effects of naphthofluorescein that were observed in immunodeficient mice seem to mainly reflect the effects of Mint3 inhibition in cancer cells. However, the suppression of tumour growth of MDA-MB-231 cells by naphthofluorescein was moderate compared with the results in a previous report on Mint3 depletion in MDA-MB-231 cells18, which implies insufficient concentration and persistence duration of naphthofluorescein within tumour tissues. In addition, naphthofluorescein is less water-soluble and unsuitable for oral administration. Further modifications of naphthofluorescein and/or combination with appropriate drug delivery systems might improve the efficacy of anti-tumour effects and provide other options for routes of administration, including oral administration.
Many studies have demonstrated that HIF-1 inhibition is an effective strategy for cancer therapy in various experimental models, and our results of an inhibition of Mint3-mediated HIF-1 activation via FIH-1 by naphthofluorescein parallel these reports6. In turn, PHD inhibitors have been reported to show anti-tumour effects in some experimental models47,48,49. PHD inhibitors induce the accumulation of not only HIF-1α but also HIF-2α and increase erythropoiesis via HIF-2 accumulation in the kidney50. This might improve hypoxic conditions in tumours, resulting partly in the anti-tumour effects. In addition, HIF activities must be controlled properly for tumour progression in response to the tumour microenvironment. Thus, aberrant accumulation of HIFs caused by PHD inhibitors might also show anti-tumour effects similar to those of HIF inhibition. Clinical trials on the HIF-2-specific inhibitor PT2385 and its derivative PT2977 are ongoing, whereas highly specific HIF-1 inhibitors are currently being developed51,52. HIF-1 is essential not only for cancer and inflammatory diseases but also for the homoeostasis of normal organs; HIF-1α KO mice show diverse abnormalities in various organs7,8,9,10,11,12. In addition, the protective roles of HIF-1 in cardiovascular diseases imply the contraindication of systemic HIF-1 inhibition in individuals with heart failure13. Thus, it might be difficult for HIF-1-specific inhibitors to avoid adverse effects. In contrast, Mint3 activates the transcriptional activity of HIF-1α in specific types of cells, such as cancer cells and macrophages, by suppressing FIH-1, and Mint3 KO mice showed no apparent abnormality23,24, which promises less adverse effects with Mint3 inhibitors. Naphthofluorescein suppressed tumour growth in an immunity-dependent manner and augmented the efficacy of cytotoxic anticancer drugs. Thus, Mint3 inhibitors are expected to be used not only individually, but also in combination with currently approved immunotherapy or cytotoxic reagents. In addition to its effects in cancer, naphthofluorescein showed anti-inflammatory effects in LPS-induced endotoxic shock. With regard to its suppression of macrophage hyperactivation, it is interesting to understand whether Mint3 inhibitors are available for modulating the cytokine storm in the coronavirus disease (COVID-19)53,54 and other chronic inflammatory diseases, such as rheumatoid arthritis and type-2 diabetes. We identified one compound that bound to Mint3 and weakly inhibited HIF-1 activity in cancer cells. Analogues of this compound were then evaluated and, among them, naphthofluorescein showed the highest HIF-1 suppression activity. Further analyses revealed that naphthofluorescein inhibited Mint3–FIH-1 interaction in vitro, the expression of HIF-1 target genes in cancer cells, and Mint3-dependent glycolysis in cancer cells and macrophages. The administration of naphthofluorescein suppressed tumour growth, metastasis, and endotoxic shock in vivo without apparent adverse effects. Overall, our work characterised Mint3 as a potential therapeutic target in cancer and inflammatory diseases, and provides insights into the effectiveness of novel approaches that combine Mint3 inhibitors and other therapeutic modalities.
In conclusion, we identified naphthofluorescein, which interferes with the protein–protein interaction between Mint3 and FIH-1 in vitro and suppresses tumour growth, metastasis, and LPS-induced endotoxic shock. Derivatives of naphthofluorescein and other compounds that target Mint3 might be useful therapeutic candidates for cancer and inflammatory diseases.
Methods
Compounds
For in vitro experiments, naphthofluorescein and other compounds for the chemical array screening were provided by RIKEN NPDepo (Saitama, Japan). For in vivo experiments, naphthofluorescein was synthesised by the Sundia MediTech Company (Shanghai, China) at a purity > 95%. Furin inhibitors I and II were purchased from Merck (Kenilworth, NJ, USA).
Chemical array screening
The chemical arrays were prepared by a previously described method55. We used 23,275 compounds from RIKEN NPDepo for the chemical arrays. Chemical array screening was conducted as previously described56,57. We used a 4-μM concentration of His6-Mint3-mCherry in the chemical arrays.
Cell culture
B16F10 melanoma and LLC cells were obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Miyagi, Japan). HT1080, MDA-MB-231, and AsPC-1 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). HT1080 cells expressing FLAG-tagged FIH-1 or V5-tagged Mint3 were established using pLenti6 lentiviral vectors (Thermo Fisher Scientific) as previously described28,58. Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; HT1080, MDA-MB-231, B16F10, and LLC) or RPMI1640 medium (AsPC-1; both from Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2. shLacZ- and shFIH-1-expressing MDA-MB-231 cells were prepared as previously described18. The sequences of shRNAs are listed in Table 1. Bone-marrow-derived macrophages were obtained by previously described methods27. All cell lines were routinely tested to exclude mycoplasma contamination.
siRNA knockdown
siRNA knockdown was carried out using Lipofectamine RNAiMAX (Thermo Fisher Scientific) as described earlier59. The siRNA sequences for each gene are described in Table 1.
Reporter assay
The reporter assay was performed as previously described, but with minor modifications14,21. A pGL4.35 reporter vector containing the firefly luciferase gene under the control of a transcriptional regulatory unit comprising 9× Gal4-binding elements was purchased from Promega. A pRL vector expressing Renilla luciferase (Promega) served as an internal control. The HT1080 cells (1 × 106) were seeded in a 90-mm dish and co-transfected with reporter plasmid (2.5 μg), internal control vector (250 ng), and pcDNA3 Gal4BD-HIF-1α CAD (727–826 amino acids) or pcDNA3 Gal4BD-HIF-2α CAD (769–870 amino acids) plasmid14,21 (250 ng) using Lipofectamine 2000 (Thermo Fisher Scientific). Twenty-four hours after transfection, the cells (1 × 104/well) were seeded in 96-well plates and treated with compounds at the indicated concentrations for 24 h. For the overexpression experiments, 50 ng/well of pcDNA3 V5-tagged Mint3, pcDNA3 V5-tagged FIH-1, or mock plasmid were transfected to HT1080 cells 2 h before compound treatment. Luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s instructions. Luminescence was measured using a GloMax 20/20 luminometer (Promega).
Preparation of recombinant proteins and GST pull-down assay
Recombinant proteins such as GST, GST-FIH-1, His6-Mint3NTmCherry, and His6-Mint3 were prepared as previously described14,18, and GST pull-down assays were performed as previously described, but with minor modifications14,18. Briefly, glutathione–Sepharose 4B (GE Healthcare)-conjugated GST fusion proteins (~1 µg) were preincubated with 0.5 mg/mL bovine serum albumin (BSA) in lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40) for 30 min at 4 °C. Thereafter, 1 µg His6-tagged FIH-1 was resuspended in lysis buffer containing 0.5 mg/mL BSA and then added to the beads. After mixing on a rotator for 2 h, the beads were washed four times with lysis buffer. The proteins were eluted with Laemmli sample buffer and analysed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by western blotting analysis.
Western blotting analysis
Cells were lysed with lysis buffer and centrifuged at 20,000 × g for 15 min at 4 °C. The supernatants were collected, and the total protein content was measured using a Bradford assay (Bio-Rad, Hercules, CA, USA). Lysates were separated by SDS-PAGE, transferred to polyvinylidene fluoride membrane filters, and analysed by western blotting. Detailed information on the antibodies used in this study is listed in Table 2. Uncropped and unedited blot images are shown in Supplementary Fig. 5.
Cell growth assay
Cells (1 × 104) were seeded into a plastic tissue culture dish and cultured at 37 °C in a humidified CO2 incubator for 5 days. The cells were counted periodically using a hemocytometer.
RNA isolation, reverse transcription, and quantitative polymerase chain reaction
Total RNA was isolated from cells or tumours using the RNeasy Plus Mini Kit (Qiagen) and subjected to reverse transcription (RT) using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). The RT products were then analysed by real-time PCR in a 7900HT qPCR system (Applied Biosystems; Foster City, CA, USA) using KOD SYBR qPCR (TOYOBO) and specific primers (Table 3) as previously described28,58. The expression levels of individual mRNAs were normalised to that of ACTB mRNA.
Measurement of lactic acid and ATP
Cells were seeded in 96-well plates (2 × 104 and 5 × 104 cells per well for MDA-MB-231 cells and macrophages, respectively) in triplicate and cultured for 24 h with or without naphthofluorescein (10 μM). For lactic acid levels, conditioned medium cultured for 6 h in fresh medium was collected, and the lactic acid contents therein were measured using an l-Lactic Acid kit (R-Biopharm, Darmstadt, Germany). For the overexpression experiments, 50 ng/well of pcDNA3 V5-tagged Mint3 or mock plasmid was transfected to MDA-MB-231 cells using Lipofectamine 3000 (Thermo Fischer Scientific) 2 h before compound treatment. ATP levels were determined using the ATP Bioluminescence Assay Kit CLS II (Roche Applied Science, Penzberg, Germany). The values obtained were normalised to protein concentrations that were determined using a Bradford Assay Kit (Bio-Rad), as previously described18,19.
Mice
Mice were maintained under specific pathogen-free conditions. Experimental protocols were approved by the Animal Care and Use Committees of the Institute of Medical Science, University of Tokyo and Institute of Medical, Pharmaceutical and Health Sciences, Kanazawa University, and the experiments were conducted in accordance with the institutional ethical guidelines for animal experiments and the safety guidelines for gene manipulation experiments. The sample size is based on the statistical analysis of variance and on exploratory experiments. BALB/c nude mice and C57BL/6J mice were purchased from Clea Japan (Tokyo, Japan). Mint3 KO mice (C57BL/6J background) have been described previously23 (Riken CLST Accession number: CDB0589K).
Toxicity test of naphthofluorescein
Eight-week-old male C57BL/6J mice were intraperitoneally injected with vehicle (saline containing 1% Cremophor EL [Sigma] and 8 mM Na2CO3) or naphthofluorescein (100 mg/kg b.w.) once daily for 5 consecutive days followed by 2 days off for 2 weeks, and the body weight of mice was periodically measured. On day 14, the mice were sacrificed, and sera, liver, kidneys, and heart were collected from each subject. Sera were subjected to biochemical tests by Oriental Yeast Co., Ltd. (Tokyo, Japan). The livers, kidneys, and hearts were fixed with 4% paraformaldehyde/PBS, dehydrated, embedded in paraffin, sliced at 4-μm thickness, and stained by ordinary hematoxylin and eosin staining prior to histological analysis.
Tumour transplantation
Six-week-old female C57BL/6J mice (E0771) and BALB/c nude mice (HT1080, MDA-MB-231, and AsPC-1) were used for the tumour growth assay using subcutaneous injection of tumour cells. For the growth studies, cells (1 × 106 for E0771, HT1080, HT1080 expressing FLAG-tagged FIH-1, and MDA-MB-231; 2 × 106 for AsPC-1) in 0.2 mL PBS were injected into the left flank of mice. On day 7 or 10, mice were randomly assigned to each experimental group. For the immunoprecipitation analysis of FLAG-tagged FIH-1 expressing HT1080 tumours, mice were injected intraperitoneally with vehicle or naphthofluorescein (100 mg/kg b.w.), and tumours were collected 3 h after injection and subjected to immunoprecipitation. For the gene expression analysis of HT1080 tumours, mice were injected intraperitoneally with vehicle or naphthofluorescein (100 mg/kg b.w.), and tumours were collected 6 h after injection and subjected to RNA isolation. For the monotherapy experiment, vehicle or naphthofluorescein (100 mg/kg b.w.) was injected into mice once daily for five consecutive days followed by 2 days off treatment. For the combination therapy of naphthofluorescein and cytotoxic anticancer reagents, vehicle/naphthofluorescein (100 mg/kg b.w.) and vehicle/anticancer reagents (doxorubicin 2 mg/kg b.w. for MDA-MB-231; gemcitabine 20 mg/kg b.w. for AsPC-1 tumours) were injected into tumour-bearing mice twice a week. Tumour volumes were periodically measured with a caliper using the formula:
where d is the minor tumour axis and D is the major tumour axis.
For the experimental lung metastasis assay, 4 × 105 B16F10 or LLC cells in 0.2 mL PBS were injected into 6-week-old male C57BL/6J mice via the lateral tail veins. The mice were sacrificed 14 days after injection. All metastatic foci on the lungs were counted under a stereoscopic microscope.
Immunoprecipitation
Tumours were lysed in RIPA buffer (Sigma) by sonication and centrifuged at 20,000 × g for 30 min at 4 °C. The supernatants were collected, and the total protein content was measured using the Bradford assay (Bio-Rad). Lysates were subjected to immunoprecipitation using anti-FLAG M2 agarose beads (Sigma) as previously described20.
Flow cytometry
Macrophages and lung cells were isolated as previously described26. Single-cell suspensions were prepared using 40-μm cell strainers (BD Biosciences) and treated with antibody cocktails on ice for 30 min. Detailed antibody information is listed in Supplementary Table 4. The cells were washed with PBS containing 2% FBS and sorted using a FACS Aria (BD Biosciences). Data were analysed using FlowJo software (Tree Star, Ashland, OR, USA). Flow cytometric analysis strategy for the definition of inflammatory monocytes is shown in Supplementary Fig. 6.
Immunostaining
Frozen sections of tumour tissues were prepared and subjected to immunostaining using specific antibodies (Supplementary Table 4) as previously described26,58. The nuclei were counterstained with Hoechst 33342, and the sections were observed by confocal microscopy (Olympus, Tokyo, Japan). Paraffin-embedded sections were prepared as previously described28. Sections were cut at a thickness of 4 μm, deparaffinized in xylene, and rehydrated in a decreasing graded ethanol series. Antigen retrieval was carried out for CD8α immunostaining by autoclaving in 10 mM citrate buffer (pH 9.0) for 10 min at 121 °C, followed by cooling for 20 min. After blocking endogenous peroxidase activity with a 3% aqueous H2O2 solution for 5 min, the sections were incubated with rabbit monoclonal anti-mouse CD8α (1:200; # 98941; Cell Signaling Technology) at 4 °C overnight. After washing with Tris-buffered saline, CD8α antibodies were detected using the DAKO Envision+ Dual Link System (Agilent, Santa Clara, CA, USA). 3,3ʹ-Diaminobenzidine tetrahydrochloride was used as a chromogen, whereas hematoxylin was used as a light counterstain. Immunostaining of the cells was performed using specific antibodies (Table 2) as previously described58. Cells were counterstained with Hoechst 33342, washed five times with PBS, mounted on slides, and imaged by fluorescent microscopy (BZ-X800; Keyence, Osaka, Japan).
LPS-induced endotoxic shock model
The LPS-induced endotoxic shock model was created as previously described, with some modifications23. Briefly, mice were injected intraperitoneally with vehicle or naphthofluorescein (100 mg/kg b.w.) 24 and 6 h before the intraperitoneal LPS (10 mg/kg b.w.) injection, and their survival was analysed.
LPS-responsive cytokine production assay
For in vivo analysis, serum was obtained at the indicated time after LPS injection and subjected to ELISA using ELISA kits for mouse TNF-α, IL-6, and IL-12p70 (R&D Systems). For in vitro analysis of cytokine production, macrophages (2 × 104/well) were seeded into 96-well plates and stimulated with LPS (100 ng/mL) in the presence or absence of naphthofluorescein (10 μM) for 24 h. Supernatants of the macrophage cultures were collected, and cytokine levels were measured using ELISA kits.
CCL2 ELISA
CCL2 levels in the lungs and sera were analysed as previously described26. Briefly, mouse lungs were homogenised in lysis buffer (20 mM HEPES, 100 mM NaCl, 1% Nonidet P-40, 10% glycerol) containing Protease Inhibitor Cocktail Set II (1:100; Millipore). Lung lysates and sera were analysed with a mouse CCL2 ELISA kit (MJE00; R&D Systems).
Statistics and reproducibility
The Mann–Whitney U test, two-sided unpaired Student’s t test with Welch’s correction, and the chi-square test with Yates’ correction were used for statistical evaluations using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA), as indicated in each experiment. For Kaplan–Meier analyses, the log-rank test with Benjamini–Hochberg adjustment was performed using the R packages “survival” and “survminer.” Data are presented as the mean ± SEM or ±SD; p values < 0.05 were considered significant. Sample size is based on statistical analysis of variance and on exploratory experiments. Each in vitro experiment was replicated at least three times successfully. Animal experiments were replicated at least twice successfully.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All of the data supporting the findings of this study are available in this article and in the supplementary information files (Supplementary Data 1).
References
Kaelin, W. G. Jr. & Ratcliffe, P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 (2008).
Keith, B., Johnson, R. S. & Simon, M. C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22 (2011).
Majmundar, A. J., Wong, W. J. & Simon, M. C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 (2010).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 (2003).
Lee, P., Chandel, N. S. & Simon, M. C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 (2020).
Semenza, G. L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625–634 (2010).
Cowburn, A. S. et al. HIF isoforms in the skin differentially regulate systemic arterial pressure. Proc. Natl Acad. Sci. USA 110, 17570–17575 (2013).
Cramer, T. et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 (2003).
Iyer, N. V. et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 12, 149–162 (1998).
Rezvani, H. R. et al. Loss of epidermal hypoxia-inducible factor-1alpha accelerates epidermal aging and affects re-epithelialization in human and mouse. J. Cell Sci. 124, 4172–4183 (2011).
Ryan, H. E., Lo, J. & Johnson, R. S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 17, 3005–3015 (1998).
Takubo, K. et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7, 391–402 (2010).
Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012).
Sakamoto, T. & Seiki, M. Mint3 enhances the activity of hypoxia-inducible factor-1 (HIF-1) in macrophages by suppressing the activity of factor inhibiting HIF-1. J. Biol. Chem. 284, 30350–30359 (2009).
Sakamoto, T. & Seiki, M. Integrated functions of membrane-type 1 matrix metalloproteinase in regulating cancer malignancy: beyond a proteinase. Cancer Sci. 108, 1095–1100 (2017).
Okamoto, M. & Sudhof, T. C. Mint 3: a ubiquitous mint isoform that does not bind to munc18-1 or -2. Eur. J. Cell Biol. 77, 161–165 (1998).
Tanahashi, H. & Tabira, T. X11L2, a new member of the X11 protein family, interacts with Alzheimer’s beta-amyloid precursor protein. Biochem. Biophys. Res. Commun. 255, 663–667 (1999).
Sakamoto, T., Niiya, D. & Seiki, M. Targeting the Warburg effect that arises in tumor cells expressing membrane type-1 matrix metalloproteinase. J. Biol. Chem. 286, 14691–14704 (2011).
Sakamoto, T. & Seiki, M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J. Biol. Chem. 285, 29951–29964 (2010).
Nakaoka, H. J. et al. NECAB3 promotes activation of hypoxia-inducible factor-1 during normoxia and enhances tumourigenicity of cancer cells. Sci. Rep. 6, 22784 (2016).
Sakamoto, T. et al. Hypoxia-inducible factor 1 regulation through cross talk between mTOR and MT1-MMP. Mol. Cell Biol. 34, 30–42 (2014).
Koivunen, P., Hirsila, M., Gunzler, V., Kivirikko, K. I. & Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 279, 9899–9904 (2004).
Hara, T. et al. Deletion of the Mint3/Apba3 gene in mice abrogates macrophage functions and increases resistance to lipopolysaccharide-induced septic shock. J. Biol. Chem. 286, 32542–32551 (2011).
Ho, A. et al. Genetic analysis of Mint/X11 proteins: essential presynaptic functions of a neuronal adaptor protein family. J. Neurosci. 26, 13089–13101 (2006).
Hara, T., Murakami, Y., Seiki, M. & Sakamoto, T. Mint3 in bone marrow-derived cells promotes lung metastasis in breast cancer model mice. Biochem. Biophys. Res. Commun. 490, 688–692 (2017).
Hara, T. et al. Control of metastatic niche formation by targeting APBA3/Mint3 in inflammatory monocytes. Proc. Natl Acad. Sci. USA 114, E4416–E4424 (2017).
Uematsu, T. et al. Mint3/Apba3 depletion ameliorates severe murine influenza pneumonia and macrophage cytokine production in response to the influenza virus. Sci. Rep. 6, 37815 (2016).
Kanamori, A. et al. Mint3 depletion restricts tumor malignancy of pancreatic cancer cells by decreasing SKP2 expression via HIF-1. Oncogene 39, 6218–6230 (2020).
Coppola, J. M., Bhojani, M. S., Ross, B. D. & Rehemtulla, A. A small-molecule furin inhibitor inhibits cancer cell motility and invasiveness. Neoplasia 10, 363–370 (2008).
Han, J., Wang, Y., Wang, S. & Chi, C. Interaction of Mint3 with Furin regulates the localization of Furin in the trans-Golgi network. J. Cell Sci. 121, 2217–2223 (2008).
Bracken, C. P. et al. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 281, 22575–22585 (2006).
Hara, T., Mimura, K., Seiki, M. & Sakamoto, T. Genetic dissection of proteolytic and non-proteolytic contributions of MT1-MMP to macrophage invasion. Biochem. Biophys. Res. Commun. 413, 277–281 (2011).
Roebroek, A. J. et al. Limited redundancy of the proprotein convertase furin in mouse liver. J. Biol. Chem. 279, 53442–53450 (2004).
Thomas, G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 (2002).
Swierczak, A. & Pollard, J. W. Myeloid cells in metastasis. Cold Spring Harb. Perspect. Med. https://doi.org/10.1101/cshperspect.a038026 (2020).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013).
Reymond, N., d’Agua, B. B. & Ridley, A. J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 13, 858–870 (2013).
Valastyan, S. & Weinberg, R. A. Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011).
Cockman, M. E. et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl Acad. Sci. USA 103, 14767–14772 (2006).
Cockman, M. E., Webb, J. D. & Ratcliffe, P. J. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann. N.Y. Acad. Sci. 1177, 9–18 (2009).
Devries, I. L. et al. Consequences of IkappaB alpha hydroxylation by the factor inhibiting HIF (FIH). FEBS Lett. 584, 4725–4730 (2010).
Zheng, X. et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc. Natl Acad. Sci. USA 105, 3368–3373 (2008).
Roebroek, A. J. et al. Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase Furin. Development 125, 4863–4876 (1998).
Chung, Y. et al. Mint3 is dispensable for pancreatic and kidney functions in mice. Biochem. Biophys. Rep. 24, 100872 (2020).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Kachamakova-Trojanowska, N. et al. HIF-1 stabilization exerts anticancer effects in breast cancer cells in vitro and in vivo. Biochem. Pharm. 175, 113922 (2020).
Nishide, S. et al. Controlling the phenotype of tumor-infiltrating macrophages via the PHD-HIF axis inhibits tumor growth in a mouse model. iScience 21, 205 (2019).
Madsen, C. D. et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 16, 1394–1408 (2015).
Maxwell, P. H. & Eckardt, K. U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 12, 157–168 (2016).
Fallah, J. & Rini, B. I. HIF inhibitors: status of current clinical development. Curr. Oncol. Rep. 21, 6 (2019).
Choueiri, T. K. & Kaelin, W. G. Jr Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 26, 1519–1530 (2020).
Zhou, T., Su, T. T., Mudianto, T. & Wang, J. Immune asynchrony in COVID-19 pathogenesis and potential immunotherapies. J. Exp. Med. https://doi.org/10.1084/jem.20200674 (2020).
Mangalmurti, N. & Hunter, C. A. Cytokine storms: understanding COVID-19. Immunity 53, 19–25 (2020).
Kondoh, Y., Honda, K. & Osada, H. Construction and application of a photo-cross-linked chemical array. Methods Mol. Biol. 1263, 29–41 (2015).
Ito, T. et al. Novel abscisic acid antagonists identified with chemical array screening. Chembiochem 16, 2471–2478 (2015).
Kawatani, M. et al. Identification of matrix metalloproteinase inhibitors by chemical arrays. Biosci. Biotechnol. Biochem. 79, 1597–1602 (2015).
Yoshino, S. et al. EXOSC9 depletion attenuates P-body formation, stress resistance, and tumorigenicity of cancer cells. Sci. Rep. 10, 9275 (2020).
Yoshino, S. et al. The ERK signaling target RNF126 regulates anoikis resistance in cancer cells by changing the mitochondrial metabolic flux. Cell Discov. 2, 16019 (2016).
Acknowledgements
We thank Hiroki J. Nakaoka, Miho Ishiura, and Mai Nakayama for providing technical support. This work was supported by a Grant-in-Aid for Scientific Research (C) (19K07659) from MEXT; a P-CREATE (Project for Cancer Research and Therapeutic Evolution; grant number JP21cm0106211) grant from The Japan Agency for Medical Research and Development; and the Kobayashi Foundation for Cancer Research (to T.S.); Grant-in-Aid for Scientific Research (C) (18K05366) and Grant-in-Aid for Scientific Research on Innovative Areas (18H05503) from MEXT (to Y.K.).
Author information
Authors and Affiliations
Contributions
T.Sakamoto and M.S. designed the study. T.Sakamoto, Y.K., and T.H. wrote the manuscript. T.S., Y.F., T.Hara., T.Hayashi, and Y.S. performed cell and mouse experiments. Y.K., K.H., T.Shimizu, and H.O. performed the chemical array screening. Y.M., J.I., S.K., and M.S. supervised the study. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Toshiro Moroishi and Caitlin Karniski. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sakamoto, T., Fukui, Y., Kondoh, Y. et al. Pharmacological inhibition of Mint3 attenuates tumour growth, metastasis, and endotoxic shock. Commun Biol 4, 1165 (2021). https://doi.org/10.1038/s42003-021-02701-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02701-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
